EL SINDROME DE ANGELMAN:
GUIA PARA LOS PADRES
Por Lynn Miller AA, BLS, MALS.
La primera edición de este informe fue realizada como el proyecto final de Maestro de Estudios Liberales en la Universidad Mary Washington en 1995. El presente texto es una segunda edición del informe, modificado para darle una mayor concisión y claridad.
Introducción
Características del síndrome
Obtener un diagnóstico
Consideraciones médicas
Cuidados cotidianos, cuidados a
domicilio
Escolaridad
Comunicación
Notas para los padres
Conclusiones
Agradecimientos
Bibliografía
Introducción
Según la Fundación del Síndrome de Angelman (ASF), el síndrome es un desorden neurológico, asociado a un retraso mental. El primer diagnóstico data de 1965, por el Doctor Harry Angelman. Este médico inglés difundió los descubrimientos que hizo con nińos que presentaban algunos síntomas, hoy característicos del síndrome de Angelman. Su publicación inicial sobre el síndrome de Angelman trataba del estudio de 150 casos. Hoy en día más de 800 casos están censados por la ASF, de los cuales, 500 están en los Estados Unidos. En el diario de la ASF, se indica que hay miles de casos no reconocidos, no diagnosticados, confundidos con el autismo, la parálisis cerebral u otras enfermedades. Hay pocas fuentes de información sobre este síndrome. Además, mucha de la información escrita está dirigida a los médicos, no a los padres. Sin embargo, los padres tienen la necesidad de leer publicaciones que, a la vez de informativas, les resulten útiles sin ser demasiado clínicas y técnicas. Por ejemplo, los padres deben saber que, salvo para el tratamiento de las crisis, no necesitan ningún tratamiento médico específico, al igual que los pronósticos más recientes indican una esperanza de vida normal para las personas aquejadas por este síndrome.
Desgraciadamente, el primer contacto de los padres con el síndrome de Angelman es, por regla general, duro y desconcertante. Cuando mi marido y yo supimos que nuestra hija estaba afectada por el síndrome, la única información que teníamos era una hoja fotocopiada que recibimos con informaciones generales sobre la enfermedad. Allí se enumeraban las capacidades y competencias que ella no podría tener jamas, o que podría alcanzar con un esfuerzo considerable. Salvo las buenas palabras de apoyo, ninguna o muy poca información práctica estaba disponible para ayudar a los padres a afrontar las dificultades de su hijo. Nuestra primera reacción (y creo que la de muchos padres), fue la tristeza, el desespero y la frustración. Sin embargo, una vez asumida esta circunstancia, decidimos orientar nuestros esfuerzos en una dirección más constructiva, recogiendo información de interés para los padres y resumiéndolos en una guía que cubriera aspectos como los síntomas característicos del síndrome. Esta guía integraría además de los diagnósticos, pronósticos y estrategias de comunicación y de educación de los nińos, incluso consejos de otros padres.
Con este objetivo, se envió un cuestionario a 288 familias de la ASF en Florida, incluyéndolo en su circular trimestral. De los 179 que llegaron cumplimentados, 85 eran nińos y 94 nińas. La edad media de los nińos era de 9 ańos, el mayor tenía 14 y el más joven 4. Sólo 6 familias declararon tener más de un nińo con el síndrome. Estos cuestionarios sirvieron de base para el primer estudio, otras informaciones nos las proporcionó la ASF, los médicos, los genetistas y terapeutas.
El Doctor Roy Smith, de la Universidad Mary Washington estableció la metodología de evaluación de los cuestionarios. Esta metodología definió un valor numérico para cada respuesta posible al cuestionario. Los datos extraídos del estudio fueron integrados en una plantilla y luego analizados estadísticamente. Los resultados fueron resumidos e incluidos en el presente documento, pensando en difundirlos a través de la ASF.
Características del Síndrome
Siendo el resultado de un sondeo, esto es una lista de especificidades comunes de comportamiento y características del síndrome de Angelman, comportamientos que no son completos dentro de la literatura clínica, pero de interés notable para los padres:
| Necesidad de contacto. | |
| Se distraen fácilmente. | |
| Comen de forma autónoma. | |
| Aprecian las fotos y los videos suyos y de su familia. | |
| Les gusta reflejarse en los espejos y en el agua. | |
| Les gusta nadar y jugar en el agua. | |
| Están dotados de buen sentido del humor. | |
| Pueden aprender un lenguaje de signos modificado, utilizando una comunicación por medio de imágenes. (La ASF presta asistencia) | |
| Poseen buena memoria para los rostros y los lugares. | |
| Tienen el sentido del tacto más desarrollado. | |
| Es muy sociable y positivo. |
Otras características mencionadas por la ASF son:
| Retraso mental importante. | |
| Ausencia de palabra. | |
| Desorden del equilibrio y de la motricidad (rigidez, movimientos bruscos, inestabilidad). | |
| A la edad de 3 ańos, la cabeza sigue siendo pequeńa. | |
| Felices, comportamiento sonriente. | |
| Prensión anormal (alrededor de dos ańos). | |
| La parte posterior de la cabeza algo achatada. | |
| Dientes pequeńos y separados. | |
| Estrabismo. | |
| Piel, cabellos y ojos claros. | |
| Piernas espaciadas, con pies planos y girados hacia el exterior. | |
| Extremidades pequeńas, talla pequeńa. | |
| Sudación excesiva, soportan mal el calor. | |
| Lengua, a menudo, salida y mandíbula prominente. | |
| Salivan y mastican excesivamente. | |
| Problemas de alimentación durante la infancia. | |
| Brazos levantados (como una muńeca). | |
| Fracaso en la posición de sentado y reptación alrededor de los 6-12 meses. |
Obtener un diagnóstico
Los nińos aquejados por el síndrome de Angelman son diagnosticados, a menudo, entre los 3 y 7 ańos por un neurólogo o genetista. Se constata una deleción en el cromosoma 15 en el 50 u 80 % de los casos. Los sondeos confirman esta constatación, como se puede ver en la ilustración 1.
| Según el genetista Joann Bodurtha de la Universidad de Medicina de Virginia, la deleción es aleatoria, y no se suele trasmitir. En aquellos casos en los que no hay una deleción, el cromosoma está modificado, lo que hace pensar en que los padres son los portadores potenciales. Hay, por tanto, un 25 % de posibilidades de que otro hijo pueda estar afectado por el síndrome. Esta conclusión se confirma también con el presente estudio. | 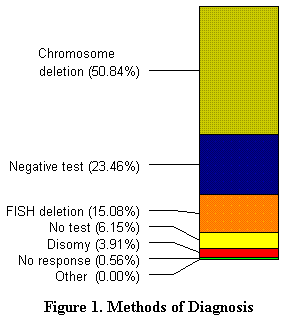 |
Sobre 6 familias que tenían mas de un hijo con el síndrome, todas indicaron que en sus informes médicos se mencionaba la ausencia de ruptura en los cromosomas.
Para los padres que se plantean tener otros hijos, el hecho de saber si hay o no deleción en el cromosoma es de vital importancia. Por eso se les aconseja hacerse un estudio cromosomático que permita a los padres entrar en contacto con el ginetista con quien podrán llevar a cabo un estudio de las posibilidades de tener otros hijos.
En diferentes universidades de los Estados Unidos y en Centros de Investigación, hay estudios en curso sobre las formas de transmisión del síndrome de Angelman.
Consideraciones médicas
Los problemas médicos giran, principalmente en torno a la gestión de las crisis. Sobre las 179 encuestas, el 75 % de los nińos tienen crisis. Como se indica en la ilustración 2, casi la mitad de los nińos que sufren crisis, estaban tratados con Depekote (ácido valproico). Los otros tratamientos posibles son a base de Klonopin, Phenobarbital o combinaciones de varios medicamentos.
| Encontrar el buen medicamento o la buena combinación de medicamentos puede resultar difícil, hacerlo de forma que el nińo lo tome también puede resultar problemático. El medicamento se suele administrar por vía oral, directa o indirectamente. La forma directa más fácil es utilizar una jeringa o un cuentagotas. | 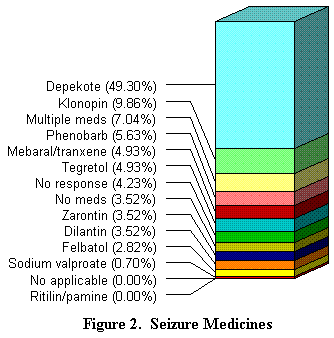 |
El método indirecto más extendido (entre el 55 % de los encuestados), es esconder el medicamento en la comida, por ejemplo, metiendo partículas de Depekote en el helado o los pasteles. Sin embargo, este método puede presentar aspectos negativos. Por ejemplo, un padre contaba que, cuando su hijo era más joven, rechazaba, uno tras otro, todos aquellos alimentos en los que se le había escondido el medicamento. Finalmente, cuando el nińo creció, se hizo menos difícil.
A parte de las crisis, no parecen existir otros males relacionados con el síndrome de Angelman. Sin embargo, como lo sugieren muchos padres con sus comentarios, el hecho de que los nińos tengan dificultades a la hora de toser y sonarse, facilita la evolución de catarros en neumonías o en otras infecciones respiratorias.
Cuidados cotidianos, cuidados a domicilio
La documentación y los estudios disponibles indican que los nińos con síndrome de Angelman son dependientes durante toda la vida. Los cuidados cotidianos, la integración escolar y un método de comunicación pueden mejorar su independencia.
La independencia para los padres es también muy importante. Tanto como los cuidados cotidianos. Una absoluta necesidad. Sobre el 84 % de las familias que pueden acceder a los servicios de ayuda a domicilio, el 65 % recurren a él. Los auxiliares a domicilio y las agencias representan el 43 % de los cuidados a domicilio, el resto cubre esta necesidad con la familia y los amigos (ver figura 3).
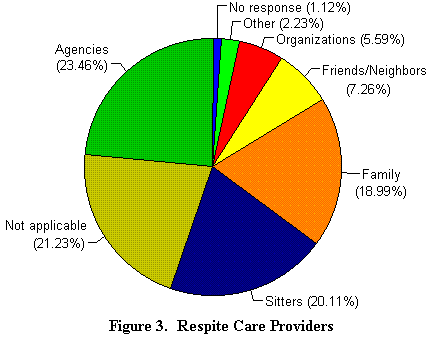
Cualquiera que sea la localización, los padres debes pagar entre 3 y 10 dólares americanos cada hora por el cuidado a domicilio (según la encuesta). 14 de entre ellos indican que cubren estos costes con mutuas privadas o con la seguridad social. Los cuidados a domicilio también implican cierto número de adaptaciones, como se indica en la ilustración 4, por ejemplo cubrir los enchufes, bloquear la apertura de armarios, proteger la habitación del nińo, cerrar las puertas, utilizar semi-puertas para la cocina, invertir las cerraduras de las puertas de las habitaciones (todo son sugerencias extraídas de la encuesta). Sin embargo, cuando los nińos crecen, estas precauciones dejan de ser necesarias, aunque ellos siguen necesitando una atención continuada.
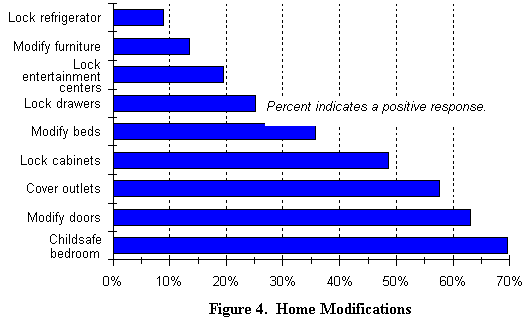
Escolaridad
Los nińos con síndrome de Angelman, requieren un curso y una clase flexible. Se necesitan asistentes que ayuden al profesor y que aseguren una vigilancia y aprendizaje normales. El 34 % de los nińos implicados en este estudio, reciben una escolaridad específica, integrándose, cuando es posible, en las clases normales. El 31 % siguen una escolaridad completamente adaptada y el 13 % están totalmente integrados en las clases normales. 4 familias proporcionan a sus hijos la enseńanza a domicilio. 6 están en colegios privados, 12 cuyos nińos son más mayores están en residencias colectivas. 2 familias han indicado que recurren a enfermeras por otras afecciones y 7 nińos no tenían la edad de ir al colegio cuando se realizó la encuesta. En total, el 85 % de los nińos van al colegio público (ver ilustración 5).
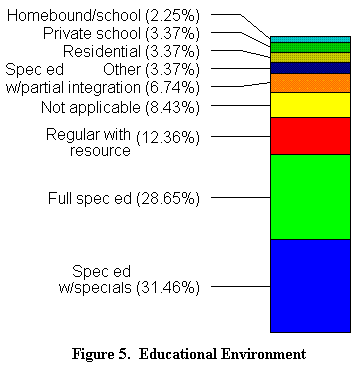 |
En el dispositivo de la escuela pública, un equipo de animadores, médicos y logopedas, ayudan a seguir el plan pedagógico personal. Los ergoterapéutas ayudan a comer y a la motricidad fina. Los psicomotricistas y los fisioterapeutas se ocupan de la ambulación, la locomoción, ayudan a levantar los pies o cambiar de posición. |
Los logopedas se concentran en la comunicación verbal o no verbal. Todos estos terapeutas, así como los asistentes, participan en los esfuerzos del profesor sobre el comportamiento del nińo y permiten una cierta independencia en los juegos y actividades de la vida diaria. Jill Clayton-Smith seńaló en su estudio de 82 pacientes en Inglaterra, que los comportamientos destructivos y la falta de sueńo fueron mejorando con terapias del comportamiento. En consecuencia, un trabajo consistente y permanente sobre el comportamiento en la escuela y en el domicilio, permiten al nińo ir al bańo sólo y desarrollar su propia destreza.
Comunicación
La comunicación es, probablemente, el elemento más vital en el pronóstico de la evolución del síndrome de Angelman, ya que es la clave de la comprensión de las capacidades cognostivas del nińo.
El estudio revela una gran disparidad (Ilustración 6) en las capacidades cognostivas evaluadas.Los padres perciben, con frecuencia, mejor, el potencial cognitivo del nińo del hecho del sistema de comunicación no verbal que desarrollan. Un padre escribe: La ausencia de palabra, el hecho de que el no pueda ir al bańo sólo, sus problemas para andar, hacen que se le considere profundo. Sin embargo, él comprende más de lo que puede expresar. |
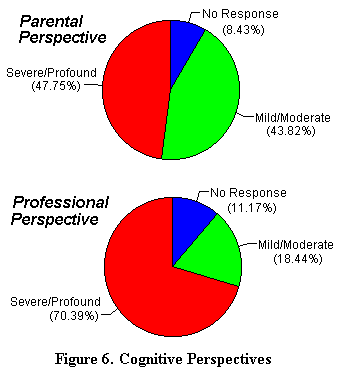 |
Los profesionales pasan un tiempo limitado con el nińo y, por tanto, tienen mayores dificultades a la hora de evaluar sus capacidades. Esto es debido al hecho de que ellos utilizan la capacidad verbal como medida de la capacidad cognitiva; mientras que los padres utilizan técnicas de comunicación no-verbal. Los dos métodos deben ser combinados para comprender el total de las capacidades cognitivas del nińo.
Si el 88 % de los nińos no pueden hablar, żcómo se pueden comunicar?. Como muestra la Ilustración 7, la mayoría de los nińos se comunican de forma íntima utilizando sonidos, movimientos de manos, gestos, moviendo su cabeza para decir si y no, utilizando el movimiento de los ojos y otros lenguajes corporales.
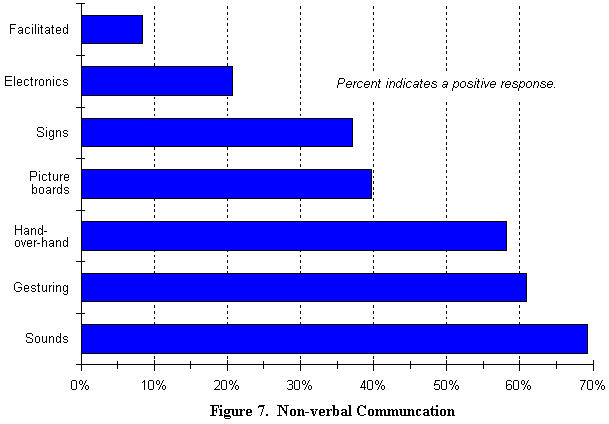
Sin embargo, la comunicación electrónica se potencia en el entorno escolar (son comentarios extraídos del estudio). También se les anima a comunicarse vocalmente a partir del momento en que ellos comprenden lo que se dice.
El proceso pedagógico debe seguir estas grandes líneas:
| Facilitar el juego potenciando la imitación y / o el juego independiente. | |
| Animar al nińo a prestar atención utilizando sonidos hablados y no hablados. | |
| Acentuar el seguimiento del comportamiento. | |
| Animar el desarrollo de la palabra y del comportamiento. |
Barbara Armstrong también indicaba que los juguetes eléctricos son de gran ayuda, no sólo por la comunicación, sino también por el juego independiente. La revista "Padres excepcionales" menciona otras informaciones sobre la comunicación, importantes para los padres y los terapeutas, particularmente en la página 36 del número de noviembre/diciembre 1992 (que incluye una lista de fabricantes de productos de iniciación), y el total del número de enero de 1995 en el que figuran las direcciones y números de teléfono de todos los organismos y empresas a escala nacional, relativos a los padres con nińos distintos.
Notas para los padres
Este estudio muestra que el 65 % de los padres son optimistas en cuanto al futuro de su hijo. Confirmando el estudio de Barbara Armstrong, las familias indican que, a través de las modificaciones del comportamiento, sus hijos pueden progresar y aprender técnicas como comer, bańarse, vestirse, jugar, ir de compras, cocinar, realizar tareas del hogar y viajar.
Una familia presenta así su filosofía sobre los medios para ayudar a su hijo:
"El 90 % de la información nos llega de un experto: nuestro hijo. Tiene el síndrome de Angelman y sabe lo que puede o no hacer. Nosotros no le impedimos nada, nosotros le educamos. Nosotros no escuchamos a los que nos dicen que él no puede, no debería o no hará."
El estudio ha posibilitado también el conocer otras preocupaciones de los padres tales como las vacaciones, las formas de distracción, las preferencias culinarias y los juguetes preferidos. Incluso si esos temas no se tratan en las documentaciones clínicas, son de suma importancia para los padres, porque les ofrecen alternativas que permiten que la vida cotidiana sea más fácil. Por ejemplo, ciertas personas entrevistadas han dicho que jamas cogían vacaciones. Sin embargo, aquellas que sí las toman, prefieren alquilar una casa cerca de un lago o de una playa.
El viaje a Disneyland es otro destino que se mencionó, con frecuencia, en la última convención del síndrome de Angelman como favorable. También se sugirieron excursiones de un día, ya que los nińos adoran viajar en coche. Las formas de distracción posibles son, por ejemplo, los libros interactivos, balones, juguetes musicales, todo lo que hace ruido, canciones, televisión, parques de atracciones, comer en el exterior, ir al estadio, hacer las compras, natación, bicicleta, pescar, hacer bowling, ir en barco, hacer visitas, columpiarse, escuchar música. Incluso al llegar a la veintena o treintena, a los pacientes con síndrome de Angelman las canciones y el mimo.
En lo relativo a la alimentación, todo es aceptable, salvo la carne ( esto depende de la elección de las familias).
Los juguetes preferidos son los libros, los juguetes de plástico que puedan llevarse a la boca, los lego, puzzles, espejos, juguetes apilables y cambiantes, juguetes eléctricos. Cuando son más mayores, según los comentarios anexos al estudio, sus gustos televisivos se amplían para incluir las emisiones prácticas, las comedias musicales, las comedias y los reportajes de animales. Las familias informan que sus hijos rechazan las películas en las que los personajes resultan heridos, lloran o están tristes. Los nińos ríen y pasan buenos momentos viendo una comedia. Según los padres, les gustan los espectáculos felices y las películas como: El sonido de la música, ET, El mago de Oz, y emisiones como Barney, Barrio Sésamo.
Conclusiones
Globalmente, los resultados de la encuesta completan las informaciones dadas por los doctores, genetistas y la ASF sobre temas como las características, diagnósticos, tratamientos y pronósticos. El estudio ha permitido, además, un acercamiento a la comunicación, demostrando que los métodos verbales y los no – verbales de educación, ayudan, ambos a que los padres y médicos puedan comunicarse con el nińo.
Uno de los aspectos que no se han tratado en la encuesta es la pubertad. Este tipo de información todavía no está disponible y para tenerla, será necesario llevar a cabo una investigación y estudio más profundos.
Finalmente, el síndrome de Angelman es un diagnóstico bastante reciente, sobre el que se están realizando avances en la investigación. Nosotros sugerimos a los padres que permanezcan en contacto con la Fundación sobre el Síndrome de Angelman (ASF) de Florida, apartado de correos 12437, Gainesville, 32604 Florida _ USA, así como con otras familias implicadas. La ASF tiene un papel clave a la hora de mantener en contacto a los padres con los investigadores del síndrome, teniendo en cuenta que son pocas las familias afectadas y pocas las instituciones que trabajan en la investigación. El apoyo y la información continuos son necesarios.
Agradecimientos
Yo tengo previsto continuar estudiando el síndrome de Angelman desde el punto de vista de los padres y, con los resultados, actualizar este informe. Actualmente, la edad media de los nińos aquejados es de 10 ańos. Cuando crezcan, yo desearía conocer y transmitir información de interés para otras generaciones. Algunos padres solicitan información sobre la pubertad. Este puede ser el objeto de mi próximo trabajo. Ocurra lo que ocurra, agradezco a la Universidad Mary Washington su ayuda en la investigación, al Doctor Roy Smith por su experiencia sobre las encuestas y por la coordinación del proyecto, al Doctor Janine LaSalle por las informaciones sobre la genética, a Jill Hendrickson de la ASF por sus sugerencias y su ayuda, a todas las familias que han respondido al cuestionario y a mi marido por su amistad y su amor infinitos.
Bibliografía
- "Angelman Syndrome: A Parent's Guide Survey."
- (179 returned surveys from parents caring for children diagnosed with AS) Fredericksburg: Mary Washington College, Masters of Arts in Liberal Studies Program, 1995.
- Angelman Syndrome Foundation.
- Angleman Syndrome. (An informational brochure made possible by contributions to the March of Dimes Birth Defects Foundation and its Campaign for healthier babies) Gainesville: Angelman Syndrome Foundation, 1994.
- Armstrong, Barbara.
- "Angelman's Syndrome: Augumentative/Alternative Communication."
- Communication Outlook. Spring 1992, 4-21.
- Bodurtha, Joann M.D., M.P.H.
- Clinical Genetics. (Clinical report) Richmond: Medical College of Virginia--Virginia Commonwealth University, 1991.
- Clayton-Smith, Jill.
- "Clinical Research on Angelman Syndrome in the United Kingdom: Observations of 82 Affected Individuals." American Journal of Medical Genetics. Manchester, England: Wiley-Liss, Inc., 1993, 46:12-15.
- Penner, Kandace A., Joy Johnston, Barbara H. Faircloth, Patricia Irish, and Charles A. Williams.
- "Communication, Cognition, and Social Integration in the Angleman Syndrome." American Journal of Medical Genetics. Gainesville: Wiley-Liss, Inc., 1993, 46:34-39.
- Waaland, Pamela K. Ph.D.
- Neuropsycological Evaluation. (Case Study Report) Richmond:Pamela Waaland, Licensed Clinical Psychologist (written evaluation of Angelman Syndrome child at age 6 years 11 months, 2 December 1992.)
- Williams, Charles M.D.; Jill Hendrickson, M.S., M.S.S.W.; Elaine Whidden, M.S.N., A.R.N.P.; Beth Buehler, M.S.
- Facts About Angelman Syndrome. (22 page report) Gainesville: Raymond C. Phillips Research and Education Unit, Division of Genetics, Department of Pediatrics, University of Florida, 1992.
- Zackowski, J.L.; R.D. Nicholls; B.A. Gray; A. Bent-Williams; W. Gottlieb; P.J. Harris; M.F. Waters; D.J. Driscoll; R. T. Zori and C.A. Williams.
- "Cytogenetic and Molecular Analysis in Angelman Syndrome." American Journal of
Medical Genetics. Gainesville: Wiley-iss, Inc., 1993, 46:7-11.
- Zori, Roberto T. M.D.; Jill Henderickson, M.S.; Sheila Woolven; Elaine M. Whidden, M.S.N., A.R.N.P.; Brian Gray, M.S.; Charles A. Williams, M.D.
- "Angelman Syndrome: Clinical Profile." Journal of Child Neurology. vol. 7 July 1992, 270-280.
Investigaciones posteriores
- Clayton-Smith, Jill and M.E. Pembrey.
- "Angelman Syndrome." Journal of Medical Genetics. 1992, 29: 412-415.
- Knoll, Joan H.M., Robert D. Nicholls, and Marc Lalande.
- "On the Parental origin of the deletion in Angelman Syndrome." Human Genetics. 1989, 83:205-206.
- Lalande, Marc.
- Angelman Syndrome and Parental Imprinting of Human Chromosome 15. (Report) Boston: Harvard Medical School, 1993.
- ----------------.
- "Imprinting and human genetics." Current Science. 1992, 4:978-982.
- ----------------, Joseph Wagstaff and Joan H. M. Knoll.
- "Molecular Analysis of Angelman and Prader-Willi Syndromes." Genome Research in Molecular Medicine and Virology. Boston: Academic Press, Inc., 1993.
- ---------------, Joseph Wagstaff, Daniel Sinnett, Valerie Greger, and Joan H. M. Knoll.
- "Mapping of the Angelman and Prader-Willi Syndromes." The Phenotypic Mapping of Down Syndrome and other Aneuploid Conditions. Boston: Wiley-Liss, Inc., 1993, 225-234.
- Lierde, Andrea Van, Maria Gabriella Atza, Daniela Giardino, and Francesco Viani.
- "Angelman's Syndrome in the First Year of Life." Developmental Medicine and Child Neurology. 1990, 32:1011-1015.
- Malcolm, S., J. Clayton-Smith, M. Nichols, S. Robb, T. Webb, J.A.L. Armour, A.J. Jeffreys, and M.E. Pembrey.
- "Uniparental Paternal Disomy in Angelman's Syndrome." The Lancet. vol 337. 23 March 1991, 694-697.
- Wagstaff, J., J.H.M. Knoll, K.A. Glatt, Y.Y. Shugart, A. Sommer and M. Lalande.
- "Maternal but not paternal transmission of 15q11-13--linked nondeletion Angelman Syndrome leads to phenotypic expression." Nature genetics. vol 1, July 1992, 291-294.
- Williams, Charles.
- Angelman Syndrome. (2 page initial report) Gainesville: Department of Pediatrics, University of Florida., 1991.
- Yamada, Kelvin A. and Joseph J. Volpe.
- "Angelman's Syndrome in Infancy." Developmental Medicine and Child Neurology. 1990, 32:1005-1010.
![]()