 Realidades
sobre el Síndrome de Angelman
Realidades
sobre el Síndrome de Angelman
Información para Familias
National Angelman Syndrome FoundationPost Office Box 12437
Gainesville, Florida 32604
U.S.: 1-800-432-6435
Outside U.S.: 212-448-1391
Indice de Contenidos
Convulsiones
Forma de andar y problemas de movimiento
Hiperactividad
Risa y felicidad
Habla y lenguaje
Retraso mental y pruebas de desarrollo
Hipopigmentación
Estrabismo y albinismo ocular
Estructura del Cerebro
Trastornos del sueńo
Problemas de alimentación y conductas motórico-bucales
Crecimiento físico
Educación
Adolescencia
Pruebas de laboratorio
Consejo genético
Reconocimientos
Referencias
Recursos en Internet:
 |
En 1965, el Dr. Harry Angelman, un médico inglés, describió por primera vez a tres nińos con características, ahora conocidas, como el Síndrome de Angelman (AS)1 . Él notó que todos tenían rigidez, andar espástico, ausencia de habla, risa excesiva y crisis convulsivas. |
Otros casos fueron publicados2,8 de forma esporádica pero la característica fue considerada sumamente rara y muchos médicos dudaron de su existencia. Los primeros informes de América del Norte aparecieron a principios de 1980s9-10 y dentro de los últimos cinco ańos han aparecido muchos informes nuevos.11,15 El Dr. Angelman relata lo siguiente con respecto a su descubrimiento de este syndrome16
"La historia de la medicina está llena de historias interesantes sobre el descubrimiento de enfermedades. La saga del Síndrome de Angelman es una de esas historias. Fue por pura casualidad que hace casi treinta ańos, tres nińos impedidos, fueron admitidos en varias ocasiones a mi centro de cuidado de nińos en Inglaterra. Ellos tenían una variedad de discapacidades y aunque a primera vista parecían estar padeciendo afecciones diferentes, yo tenía la sensación que había una causa común para su enfermedad. El diagnóstico fue puramente clínico porque a pesar de investigar sobre las pruebas físicas existentes, las cuales hoy en día son mucho más refinadas, no fui capaz de establecer, con una prueba científica, que los tres nińos tenían la misma discapacidad. En vista de esto dudé de publicar mis estudios sobre ellos en revistas médicas. Sin embargo, estando de vacaciones en Italia, vi una pintura al óleo en el museo de Castelvecchio en Verona llamada . . un Muchacho con una muńeca. La cara de sonrisa del muchacho y el hecho de que mis pacientes mostraran movimientos rígidos, me dieron la idea de escribir un artículo sobre los tres nińos con el título de Nińos Muńeca. No fue un nombre que agradara a todos los padres pero sirvió como un medio de incluir a los tres pequeńos pacientes en un solo grupo. Después el nombre se cambió al síndrome de Angelman. ' Este artículo se publicó en 1965 y después de un cierto interés inicial casi se olvidó hasta los primeros ochenta."
La incidencia exacta del AS es desconocida pero en los Estados Unidos y Canadá, la 'Angelman Syndrome Fundation' tiene conocimiento de la existencia de aproximadamente 1000 individuos, así que el síndrome no es sumamente raro. Casos de AS se han reportado a lo largo del mundo entre grupos raciales distintos. En América del Norte, la gran mayoría de casos conocidos parecen ser de origen Caucásico. La incidencia exacta de AS es desconocida y estimaciones de un 1 caso por cada 15,000 - 30,000 nacimientos parecen razonables.
Características físicas y de desarrollo
El síndrome de Angelman, normalmente, no se reconoce en el recién nacido o en la
infancia, dado que los problemas de desarrollo son inespecíficos durante este tiempo. Los
padres, en primera instancia, pueden sospechar el diagnóstico después de leer sobre AS o
ver a un nińo de esas características. La edad más común de diagnóstico está entre
tres y siete ańos cuando las conductas características y rasgos se hacen mas evidentes.
Un resumen del desarrollo y las características físicas ha sido recientemente publicado17
con el propósito de establecer criterios clínicos para el diagnóstico y éstos están
relacionados mas abajo. Todos los rasgos no necesitan estar presentes para que el
diagnóstico pueda ser hecho y el diagnóstico, a menudo, es lo primero que se sospecha
cuando las conductas típicas son reconocidas.
Desarrollo evolutivo y pruebas de Laboratorio
Estrabismo |
Hipopigmentación de piel y ojos |
Lengua prominente; problemas para succionar/tragar |
Hiperactividad de movimientos reflejos en tendones |
Problemas con la alimentación durante la infancia. |
Brazos levantados y flexionados al caminar. |
Mandíbula prominente |
Hipersensibilidad al calor |
Boca grande, dientes espaciados |
Problemas para dormir |
Babeo frecuente, lengua fuera. |
Atracción hasta la fascinación por el agua |
Conductas excesivas en mascar/masticar. |
Aplastamiento posterior de la cabeza. |
Cromosoma 15
Durante varias décadas el estudio del cromosoma de AS no reveló ninguna anormalidad
pero, con el desarrollo de nuevos métodos de análisis, se encontró, en el cromosoma 15,
que faltaba un área muy pequeńa . Los más recientes métodos de análisis moleculares
demuestran que existe una deleción en aproximadamente 70% de individuos con AS. El área
anulada, aunque sumamente pequeńa, es realmente bastante grande cuando se analiza a nivel
molecular. Se cree que tiene casi 3.5 millones de moléculas de longitud, bastante
distancia como para contener muchos genes.
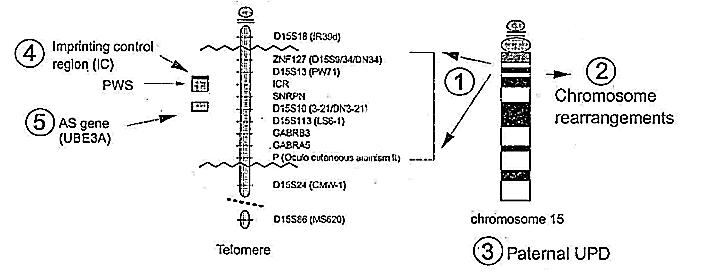
La región anulada en el cromosoma 15 se sabe que contiene genes que están activados o
desactivados dependiendo del origen materno o paterno del cromosoma (el cromosoma 15
heredado de la madre tiene el gen activado pero el mismo gen heredado del padre está
desactivado). Dado que las delecciones vistas en AS sólo ocurren en el cromosoma 15
heredado de la madre, se cree que el gen sólo se activa en el cromosoma materno. Ningún
gene/s AS ha sido aislado todavía aunque esto puede ocurrir pronto. La deleción en genes
que están activados y que son de origen paterno se sabe que causan otro desorden de
retraso mental conocido como el síndrome de Prader-Willi (PWS). Los gene/s de PWS
realmente se localizan cerca del gen AS, pero son diferentes.
Después del descubrimiento de la deleción en el cromosoma han sido descubiertos otros
casos raros de AS. Estos casos eran debidos a que el nińo tenía los dos cromosomas 15
heredados del padre, y se les ha dado el nombre de Uniparental Disomía (UPD). En estos
casos no existe deleción pero el chico ha perdido el gen AS activado ya que los
cromosomas 15 heredados del padre sólo tienen genes no activados.
Además, hay también algunas familias que tienen dos o más hijos con AS. En muchas de
estas familias se ha visto que los nińos AS siempre heredaban el mismo cromosoma 15 de la
madre pero heredaban diferente cromosoma 15 del padre indicando esto que el cromosoma
materno heredado puede tener una mutación en él. Han sido identificados dos
mecanismos genéticos a través de los cuales el gen AS puede ser heredado de la madre:
Mutaciones en la región del centro de control de 'Imprinting' (lugar donde se activan los
genes maternos) y mutaciones en el supuesto gen AS el cual ha sido denominado como
'Ubiquitin-protein ligase E3A (UBE3A)'.18,19 El gen UBE3A se cree que es
el causante de AS, y todos los otros mecanismos genéticos que están asociados con AS
(ver más abajo), aparecen como consecuencia de que ese gen no esté activado o no esté
presente. UBE3A es un componente enzimático de un sistema de degradación de una
compleja proteína llamado 'the ubiquitin-proteasome pathway'.20
This pathway (camino) esta situado en el citoplasma de todas las células. Este camino
implica la actividad de una pequeńa molécula proteínica, 'ubiquitin', que se puede unir
a otras proteínas causando de este modo su degradación. No obstante, por el momento, no
sabemos a que proteínas del cerebro degrada la encima UBE3A.
Se sabe también que hay una región en el cromosoma 15 que puede
controlar la activación o desactivación de la acción del gen UBE3A. Esta región
de control se denomina 'Imprinting Center' (IC) y se han identificado pequeńas mutaciones
en este área que pueden causar AS.21 El IC parece ser capaz de ejercer su
efecto en UBE3A desde una localización lejana pero no se sabe todavía como se produce
este mecanismo de actuación.
Todos estos descubrimientos han llevado a saber que existen varios
'tipos' de mecanismos genéticos que producen AS y todos, generalmente, conducen a las
típicas características clínicas observadas en AS, no obstante se pueden producir
pequeńas diferencias entre distintos grupos. Estos mecanismos se describen en el diagrama
de abajo y se resumen en la tabla.
Grupos Genéticos en el Síndrome de Angelman
| 1 | Grandes delecciones | 70-75%* | Incluye la deleción del gen P (Pigmentación), luego es común la existencia de hipopigmentación |
| 2 | Otras anormalidades en cromosomas | 2% | Cambios en la ordenación de cromosomas pueden causar la ausencia de la región 15q11-13 |
| 3 | Uniparental Disomía paterna | 4% | Ambos cromosomas 15 heredados del padre; no existe cromosoma 15 de origen materno |
| 4 | Mutaciones en el 'Imprinting center' | 1% | Incidencia muy escasa |
| 5 | Mutaciones en UBE3A | 3-5% | Ultimo mecanismo identificado. Incidencia real aún no determinada |
| 6 | Desconocidos | 15% | Todos los mecanismos anteriores deberían estar descartados a través de pruebas genéticas antes de ser asignado a este grupo. |
Problemas médicos y de desarrollo
Más del 90% han reportado que tienen convulsiones pero ésta puede ser una sobrestimación porque los informes médicos tienden a estudiar los casos más severos. Menos del 25% padecen convulsiones antes de los 12 meses de edad. La mayoría tiene convulsiones antes de los 3 ańos; la incidencia en nińos mayores o en adolescentes no es excepcional.13 Las convulsiones pueden ser de cualquier tipo (de tipo motórico afectando a todo el cuerpo con sacudidas de las extremidades; ausencias que conllevan periodos breves de falta de consciencia), y puede requerir medicaciones múltiples anticonvulsivas. Las convulsiones pueden ser difíciles de reconocer o diferenciar de temblores normales del nińo, movimientos hiperquinéticos de extremidades o faltas de atención. El EEG (Electroencefalograma) típico, es a menudo más anormal de lo esperado y puede hacer pensar en actividad convulsiva cuando, de hecho, no la hay.
No hay ningún consenso acerca de la medicación anticonvulsiva óptima, pero hay modelos de uso que son más frecuentes. Las medicaciones anticonvulsivas. de uso en las crisis motóricas de carácter menor (ácido valproico, clonazepam, etc.) son prescritas más normalmente que las que se utilizan para ataques mayores (diphenylbydantoin, phenobarbital, etc.). Es preferible el uso de medicación única pero es común que las crisis continúen. Se han puesto, algunos nińos con convulsiones incontrolables, en dieta ketogenética, pero no esta claro si esto es beneficioso. Nińos con AS tienen el riesgo de ser sobretratados con medicación porque pueden confundirse sus movimiento espásticos o faltas de atención con convulsiones y pueden dar EEG anormales incluso cuando las crisis convulsivas están controladas.
Forma de andar y problemas de movimientos
Movimientos Hiperquinéticos del tronco y miembros han sido reportados en los primeros ańos19 y movimientos nerviosos o temblores pueden estar presentes en los primeros 6 meses de vida. Los movimientos voluntarios son a menudo irregulares, variando de sacudidas ligeras a movimientos toscos no coordinados que se producen caminando, comiendo, y alcanzando objetos. La consecución de las etapas normales de motricidad gruesa están retrasadas; normalmente se sientan después de los 12 meses y no andan hasta los 3 o 4 ańos.13,15
En la infancia, el nińo ligeramente dańado puede andar de forma casi normal. Puede tener andares sólo apacibles o andares a saltitos. Esto puede estar acompańado por una tendencia a echarse hacia delante. Esta tendencia a echarse hacia delante se acentúa cuando corren y, además, los brazos se mantienen levantados. Para estos nińos, el equilibrio y la coordinación no parece ser un problema mayor. Los nińos más severamente afectados pueden estar muy rígidos (como un robot) y sumamente inseguros y accidentados al caminar. A pesar de que ellos pueden gatear bastante bien pueden llegar a pararse de golpe o parecer que se vuelven nerviosos cuando permanecen de pie. Las piernas permanecen separadas y los pies son planos y girados hacia el exterior. Esto, acompańado por brazos levantados, los codos encorvados y manos giradas hacia abajo, produce la forma de andar característica del AS. Algunos nińos son tan atáxicos y afectados que no logran andar hasta que son mayores y por tanto mas capaces de compensar motoricamente su rigidez; aproximadamente el 10% no llega a caminar.23 en situaciones donde AS no se ha diagnosticado, el inespecífico diagnóstico de parálisis cerebral se da a menudo en base a la forma extrańa de andar. La terapia física es normalmente útil mejorando la deambulación y a veces puede ser necesaria la intervención quirúrgica para alinear las piernas.
La hiperactividad probablemente es la conducta más típica en AS. Se describe mejor
como Hipermotricidad con un bajo tiempo de atención. Esencialmente todos los nińos AS
tienen algún componente de hiperactividad15 y varones y mujeres parecen
igualmente afectados. Tanto de nińos como de mayores pueden tener una actividad
aparentemente incesante, constantemente guardando sus manos o juguetes en su boca,
moviéndose de un sitio a otro. En casos extremos, el movimiento constante puede causar
accidentes con contusiones y rozaduras. Conductas como agarrar, pellizcar y morder a
nińos más mayores se ha constatado que, también, pueden acentuarse por la actividad
hipermotórica. Terapias persistentes y consistentes de modificación de conducta ayudan a
disminuir o eliminar estas conductas no deseadas.
El tiempo de atención puede ser tan corto que impida la interacción social al no poder
el nińo AS captar las expresiones faciales y otras seńales sociales. En casos leves, la
atención puede ser suficiente para aprender lenguaje de signos y otras técnicas de
comunicación. Para estos nińos, programas de entrenamiento educativos y de desarrollo
son fáciles de estructurar y generalmente son eficaces. Observaciones en jóvenes adultos
sugieren que la hiperactividad disminuye con la edad. La mayoría de los nińos AS no
toman medicación para la hiperactividad aunque algunos podrían beneficiarse del uso de
medicaciones como methyiphenidate (Ritalin). El uso de agentes sedantes como
phenothiazines no esta recomendado debido a su potencia y efectos secundarios.
Risa y felicidad
No se sabe por qué la risa es tan frecuente en AS. Incluso la risa en individuos normales
no se conoce bien. Estudios del cerebro en AS, usando exploración MRI o CT no han
mostrado ningún defecto que haga pensar en un sitio para una anormal risa-inducida.
Aunque hay un tipo de convulsión asociado con la risa, llamada epilepsia risible, esto no
es lo que ocurre en AS. La risa en AS parece ser, fundamentalmente, un suceso de
expresión motórica; la mayoría de las reacciones a los estímulos, físicos o mentales,
se acompańa por risa o una risa parecida a muecas faciales. Aunque los nińos AS
experimentan una variedad de emociones, aparentemente predomina la felicidad.
La primera evidencia de esta conducta característica puede estar en el comienzo de una
temprana y persistente sonrisa a la edad de 1-3 meses. Risueńo, sonriendo entre dientes y
con sonrisa constante pronto desarrollan una risa reflexiva normal pero tienen retraso o
están reducidas conductas como arrullarse y parlotear. Mas adelante varios tipos de
expresiones faciales o conductuales caracterizan la personalidad del nińo. Unos pocos
presentan una risa verdaderamente cercana al paroxismo o contagiosa y en un estudio el 70%
presentaba "estallidos de risa" .15 Las conductas de gestos de
alegría y sensación de felicidad se producen frecuentemente. En casos raros, la
apariencia de felicidad está rozando con la irritabilidad y la hiperactividad es uno de
los rasgos de personalidad predominantes; llorar, chillar, gritar una especie de cortos
sonidos guturales pueden ser las conductas predominantes.
Habla y lenguaje
Algunos nińos AS parecen tener bastante comprensión como para ser capaces de hablar,
pero incluso en los de más alto nivel, el lenguaje conversacional no se desarrolla.
Clayton-Smith23 informaron que unos individuos hablaron 1-3 palabras, y en un
estudio de 47 individuos, Buntirix et al.15 informaron que el 39% hablaron
hasta 4 palabras, pero no se indicaba si estas palabras fueron usadas de acuerdo con su
significado. Nińos con AS causado por disomía uniparental o por delecciones sumamente
pequeńas pueden tener capacidades verbales y cognoscitivas más altas; pueden llegar a
usar de 10-20 palabras aunque la pronunciación puede ser torpe."16
La discapacidad en el habla en AS tiene una evolución algo típica. Los bebés y los
nińos jóvenes lloran menos a menudo y ha disminuido el arrullarse y el balbuceo. Una
sola palabra clara, como "mamá", puede tardar en desarrollarse alrededor de
10-18 meses pero se usa infrecuentemente e indiscriminadamente sin el significado
simbólico. A los 2-3 ańos de edad, está claro que hay un retraso en el habla pero puede
no ser evidente cuan pequeńa es su capacidad verbal; llorando y con otros arranques
verbales pueden enmascarar su déficit. A los 3 ańos de edad, los nińos AS de nivel mas
alto están comenzando algún tipo de lenguaje no-verbal. Algunos apuntan a partes de su
cuerpo e indican algunas de sus necesidades a través del uso de gestos simples, pero su
nivel de comprensión es mucho mas alto a la hora de entender y seguir órdenes. Otros,
sobre todo aquéllos con deleción grande o los muy hiperactivos. no pueden mantener su
atención lo suficiente para lograr las primeras fases de comunicación, tales como
establecer contacto visual sostenido. Las capacidades de lenguaje no verbal de los nińos
AS varía grandemente; los más avanzados son capaces de aprender algún lenguaje de
signos y usar ayudas como murales de comunicación basados en imágenes.
Retraso mental y la comprobación de
desarrollo
La comprobación de desarrollo está comprometida por la falta de atención,
hiperactividad, falta de habla y control motórico. En tales situaciones, los resultados
de la prueba están invariablemente en el rango severo de deterioro funcional. Los nińos
más atentos pueden estar en el rango moderado y una minoría puede realizar algunas
categorías, como habilidades sociales receptivas, en el rango ligeramente dańado. Según
se va sabiendo más sobre las diferentes clases genéticas de AS parece que los pacientes
con disomía uniparental tienen manifestaciones clínicas menos severas que aquéllos con
delecciones grandes.24
Se sabe que las capacidades cognoscitivas en AS son más altas que las indicadas por los
test de desarrollo. El área más llamativa donde esto es evidente está en la diferencia
entre el lenguaje comprensivo y el lenguaje hablado. Debido a su capacidad de comprender
el lenguaje, los nińos AS pronto se diferencian de otros cuadros de retraso mental
severo; los jóvenes adultos con AS son, normalmente, socialmente adaptados y responden a
la mayoría de las seńales personales e interacciones. Debido a su interés por las
personas, ellos establecen amistades que son premiadas y comunican todo un amplio
repertorio de sentimientos, enriqueciendo su relación con las familias y amigos. Ellos
participan en actividades de grupo, quehaceres de la casa y en las actividades y
responsabilidades de vivir diario. Como otros, ellos disfrutan la mayoría de las
actividades recreativas como TELEVISION, deportes, yendo a la playa, etc.
No obstante hay una amplia gama en el nivel de desarrollo que hace que no todos los
individuos con AS logren las capacidades nombradas anteriormente. Unos pocos estarán más
dańados en términos de su retraso mental y falta de atención, y éste parece el caso
sobre todo en aquéllos con dificultad para controlar las convulsiones o aquéllos con
ataxia sumamente pronunciada y problemas de movimiento. Afortunadamente, la mayoría de
los nińos con AS no tienen estos problemas severos, pero incluso para el nińo menos
dańado, la falta de atención y la hiperactividad durante la infancia a menudo da la
impresión que el deterioro funcional profundo es el único resultado posible. Sin
embargo, con un hogar seguro, intervención intensa en sus perfiles de conducta y
estimulación, el nińo AS empieza a superar estos problemas y el progreso en el
desarrollo se produce.
Hipopigmentación:
Cuando AS es causado por una deleción grande, normalmente existe una hipopigmentación de
la piel y en los ojos. Esto ocurre porque hay un gen del pigmento, localizado cerca del
gen AS que también se ha perdido. Este gen del pigmento produce una proteína (llamada
proteína P) se cree que eso es crucial en la síntesis de la melanina. Melanina es la
molécula principal para la pigmentación de nuestra piel. En algunos nińos con AS, esta
hipopigmentación pueden ser tan severa que puede llegar a sospecharse una forma de
albinismo. En aquéllos con disomía uniparental o con deleción muy pequeńa, este gen no
se ha perdido y la piel es normal y la pigmentación del ojo se ve. Los nińos AS con
hipopigmentación son muy sensibles al sol, así que el uso de protectores solares es
importante. No todos los nińos AS con pérdida del gen P tienen, obviamente,
hipopigmentación, y puede darse que sólo tengan un color de piel mas claro que el de sus
padres.
Estrabismo y albinismo ocular
Estudios de pacientes con AS demuestran que la incidencia de estrabismo se da en el 30-60
% de los casos. ' Este problema parece ser más común en nińos con hipopigmentación
ocular, dado que el pigmento en la retina es crucial para el desarrollo normal de las
ramificaciones del nervio óptico. El tratamiento del estrabismo en AS es similar al de
otros nińos: evaluación por un oftalmólogo, corrección de cualquier déficit visual, y
cuando sea apropiado, parches oculares o ajuste quirúrgico de los músculos
extraoculares. Las actividades hipermotóricas de algunos nińos AS harán que sea
difícil el uso de parches oculares y gafas.
Estructura de Sistema Nervioso Central
El cerebro en AS es estructuralmente normal aunque se han informado anormalidades
ocasionales. Los cambios mas frecuentes a nivel medio o cortical, cuando se detectan, son
atrofia cortical ligera (es decir una pequeńa disminución del espesor de la corteza
cerebral) y/o ligera disminución de mielinización (es decir las partes más internas del
cerebro parecen tener un ligero grado de disminución de materia blanca). 13,15
Algunos detallados estudios microscópicos y químicos del cerebro en AS han sido
publicados pero nosotros creemos que los resultados, generalmente, han sido inespecíficos
o el número de casos ha sido demasiado pequeńo como para poder hacer conclusiones
significantes.
Trastornos del sueńo
Los padres informan que la disminución de la necesidad de dormir y ciclos anormales de
dormir/despertarse son característicos de AS. Se han reportado perturbaciones del sueńo
en nińos AS y se ha estudiado el caso de un nińo que estando en un programa de
tratamiento de conductas, los ciclos de dormir/despertar eran anormales. Muchas familias
acondicionan el dormitorio para que sea seguro y no pueda salir el nińo, de cara a
facilitar la vigilancia y cuidado durante la noche. El uso de sedantes como chloral
hidrate o diphenyihydramine (Benadryl) puede ser útil si la vigilia interfiere en la vida
de los demás miembros de la casa. Recientemente, la administración de 0,3 mg. De
Melatonina 1 hora antes de acostarse, se ha mostrado como una ayuda en algunos nińos,
pero no debe administrarse en mitad de la noche si el nińo se despierta.25 No
obstante, la mayoría de los nińos AS no recibe medicaciones para el sueńo y aquéllos
que lo hacen normalmente no requieren mucho tiempo de uso.
Problemas con la alimentación y conductas
motórico-bucales
Son frecuentes los problemas con la alimentación pero, generalmente, no son severos y se
manifiestan temprano teniendo dificultad para chupar o tragar.13,15,22 Los
movimientos de la lengua pueden no estar coordinados con el tragar y existe una falta de
coordinación motórico-bucal generalizada. Puede haber problemas cuando empiezan la
succión y se mantienen durante el amamantamiento; la alimentación con biberón puede
resultar más fácil. La conducta de escupir con frecuencia puede interpretarse como una
forma de intolerancia a la comida o un reflujo gastro-esofágico. Las dificultades con la
alimentación, a menudo, se presentan, en primera instancia, al médico como un problema
de poca ganancia de peso o como un "problema de crecimiento". Con poca
frecuencia, el reflujo gastro-esofágico severo puede requerir cirugía.
Los nińos AS son característicos por poner todo en su boca. En la nińez, es frecuente
que se chupen las manos (y a veces el pie). Mas adelante, el procedimiento exploratorio
mas frecuente es a través de la manipulación oral y masticando. La lengua parece ser de
forma y tamańo normal, pero en 30-50%, una persistente protuberancia de la lengua es un
rasgo característico. Algunos tienen protuberancia y babeo constante mientras otros
tienen una protuberancia que sólo es notable durante la risa. Algunos nińos con
protuberancia no tienen ningún problema notable al final de la nińez (algunos parecen
mejorar después de la terapia motórico-bucal). Para el típico nińo AS con conducta de
lengua prominente, el problema permanece a lo largo de la nińez y puede persistir en la
madurez. Babear frecuentemente es un problema persistente, a menudo requiriendo el uso de
baberos. El uso de medicaciones, como scopolamina, secar la baba con frecuencia, no
produce un efecto adecuado a largo plazo.
Crecimiento físico
De recién nacidos parecen estar bien formados físicamente, pero alrededor de los 12
meses de edad se manifiesta una desaceleración de crecimiento craneal que puede
representar una microcefalia absoluta o relativa ( microcefalia absoluta significa tener
un perímetro cefálico en el 2.3 percentil más bajo). El predominio de microcefalia
absoluta varía del 88%13 al 34%12 y puede ser tan bajo como 25%
cuando los casos sin deleción también son incluidos.11 No obstante la
mayoría de los individuos AS tienen perímetro cefálico menor del percentil 25 a la edad
de 3 ańos, a menudo acompańado por un aplanamiento detrás de la cabeza. La media de
estatura es más baja que la inferior para los nińos normales pero la mayoría nińos AS
estarán dentro del rango normal. La altura final de adulto ha ido de 1,45 m a 1,78 m en
una serie de 8 adultos con AS. Los factores familiares influirán en el crecimiento dando
que padres más altos tengan nińos AS que tienden a ser más altos que la media de nińos
AS. El aumento de peso durante la infancia puede ser baja debido a los problemas con la
alimentación pero ya en la nińez temprana la mayoría nińos AS tiene una cantidad de
grasa hipodérmica casi normal. La obesidad es rara pero al final de la nińez puede
ocurrir que algunos hayan aumentado de peso.23
Educación
El severo retraso en el desarrollo en AS obliga a que se establezca un completo rango de
entrenamiento temprano y programas de enriquecimiento. Los nińos con poca estabilidad o
sin capacidad de andar también pueden obtener beneficios de la terapia física. La
terapia ocupacional puede ayudar a mejorar la motricidad fina y controlar la conducta
motórico-bucal. Pueden requerirse sillas adaptables especiales o posicionadores en varios
momentos, sobre todo para los hipotónicos o extremadamente atáxicos. Logopedia y terapia
de comunicación es esencial y debe enfocarse en los métodos de comunicación no
verbales. Las ayudas que potencien la comunicación, como fotos o murales de
comunicación, deben usarse en el momento apropiado más temprano.
Sumamente activos e hipermotóricos los nińos AS requerirán equipamientos especiales en
el aula y pueden necesitarse soportes del profesor o ayudantes que integren al nińo en el
aula. Los nińos AS con déficits de atención e hiperactividad necesitan una habitación
para expresarse ellos mismos y para "luchar cuerpo a cuerpo" con sus actividades
hipermotóricas. La distribución del aula debe estructurarse, tanto en el plano físico
como en su programa de actividades, para que el activo nińo AS pueda encajar y ajustarse
al ambiente escolar. La individualización y flexibilidad son factores importantes.
Técnicas de modificación de conductas, tanto en el colegio como en casa, pueden permitir
que el nińo AS sea entrenado en sus necesidades con el retrete (programación horaria -
entrenamiento), y también para desarrollar la capacidad de realizar él mismo la mayoría
de las tareas relacionadas con el comer, vestir y realizar actividades generales en la
casa.
Adolescencia
Durante la adolescencia, la pubertad puede estar retrasada de 1-3 ańos pero la
maduración sexual ocurre con el desarrollo normal de las características sexuales
secundarias. Un poco de ganancia de peso puede ser evidente en este periodo pero la
obesidad franca es rara. Los jóvenes adultos AS continúan aprendiendo y no se conoce que
haya un deterioro significante en sus capacidades mentales. La salud física en AS parece
ser notablemente buena. Para muchos, pueden retirarse las medicaciones para las
convulsiones al principio de la adolescencia.10,15Los individuos AS con ataxia
severa pueden perder su capacidad de caminar si no se practica el andar. Durante la
adolescencia se puede desarrollar escoliosis y es un problema sobre todo en aquéllos que
no tienen capacidad de andar. La escoliosis se trata con la puesta temprana de un corsé
ortopédico para prevenir la progresión, y una corrección quirúrgica o estabilización
pueden ser necesarias para los casos mas severos. La esperanza de vida no parece estar
acortada significativamente y nosotros tenemos noticia de una mujer de 58 ańos con AS y
conocemos muchos en su tercera o cuarta década de vida.
Pruebas de Laboratorio para detectar AS
En el nińo en el que exista la sospecha de AS, lo primero que se hace, a menudo, es un
análisis de cromosomas de alta resolución para asegurar que no se trata de ningún otro
desorden genético, dado que rasgos como retraso mental, microcefalia o convulsiones
pueden verse en otras anormalidades del cromosoma. Al mismo tiempo que el análisis del
cromosoma antes mencionado, normalmente, se hace una prueba llamada FISH (Fluorescent in
situ hybridization). Esta es una prueba recientemente desarrollada que usa marcadores
moleculares para descubrir la deleción en el cromosoma 15. Los marcadores se comparan
directamente con el cromosoma examinándose bajo un microscopio después de aplicarle unos
colorantes especiales. La prueba del FISH es con mucho superior al análisis normal del
cromosomas. El nińo con AS debe tener sus cromosomas 15 totalmente estudiados para
asegurarse que ellos son estructuralmente normales; un estudio del cromosoma de la madre
proporciona confirmación adicional de que también el cromosoma 15 materno es
estructuralmente normal. En el test de diagnóstico de AS, algunos laboratorios ahora
ofrecen el test "DNA methylation" junto con el análisis de cromosoma y la
prueba FISH. La prueba del methylation puede descubrir el tipo mas corriente de AS, es
decir la gran deleción, así como aquéllos de disomía uniparental o los de defectos en
el 'imprintig center' (IC). la Confirmación de disomía uniparental necesita ser hecha
por comprobación molecular adicional (normalmente, estudio de la secuencia paterna en el
área de IC). Aproximadamente 80-85% de individuos con AS serán diagnosticados por una
combinación de estas pruebas, pero todavía quedará un 15-20% que necesitará alguna
comprobación genética. En algunos individuos en este último grupo, quizás menos de
20%, se encontrará, no obstante, que tienen mutaciones en el gen UBE3A. En este momento
no están disponibles, análisis moleculares para UBE3A y para mutaciones en el IC pero se
están desarrollando en algunos laboratorios de investigación
Consejo genético
Aproximadamente el 70-75% de los casos de AS se producen por grandes delecciones o por
disomía uniparental. En lo que nosotros sabemos, no se ha reportado para estos grupos
casos de recurrencia; y los riesgos de recurrencia se ha estimado que están por debajo
del 1 %. El diagnóstico prenatal esta disponible a través de pruebas citogenéticas y
moleculares.
Individuos con AS debido a mutaciones en el IC pueden, tanto haber heredado esta mutación
de una madre normal como haber recibido la mutación de forma espontanea (no heredada). En
el primer caso el riesgo teórico de recurrencia es del 50%; en el otro caso (mutación
espontanea) el riesgo se cree que es menor del 1%.
Aquellos individuos AS debido a mutaciones en el UBE3A, como es el caso de mutaciones en
el IC, pueden tanto haber recibido la mutación de una madre normal como haberla adquirido
de forma espontanea. El riesgo de recurrencia se cree que debe ser del 50% en el primer
caso y menos del 1% en el segundo. Se puede hacer un diagnóstico prenatal, a través de
pruebas moleculares, siempre que se trate de mutaciones en el IC o en UBE3A que hayan sido
molecularmente caracterizadas.
En casos de AS que están asociados con un cromosoma 15 estructuralmente anormal (por
ejemplo una traslocación cromosómica) puede haber un incremento del riesgo de
recurrencia. En estos casos el riesgo de recurrencia debe estar basado en la específica
anormalidad del cromosoma y lo que se sabe sobre su recurrencia. Existe la posibilidad, en
estos casos, de diagnóstico prenatal, a través de pruebas citogenéticas o moleculares.
La estimación de riesgos de repetición es muy difícil para los individuos AS que tienen
estudios genéticos normales (es decir, no tienen ninguna de las etiologias anteriores).
Existen casos de repetición familiar en este grupo, así que está claro que el riesgo de
repetición de AS es más alto que para aquéllos con, por ejemplo, la típica deleción
grande. Hasta que se sepa más sobre este grupo, la cautela es la norma de conducta
durante el consejo genético dado que el riesgo teórico de recurrencia puede ser tan alto
como el 50% (si uno asume que mutaciones no detectadas, causantes de AS, han sido
heredadas de la madre).
Debe tenerse en cuenta que los estudios del cromosoma habituales, realizados durante el diagnóstico prenatal rutinario, se interpretan a menudo como normales en fetos AS con delecciones, dado que las anormalidades pequeńas en el cromosoma 15 no son detectadas por este tipo de estudio. Son necesarias pruebas específicas para el cromosoma 15 o estudios FISH, para el diagnóstico prenatal en casos donde la comprobación busca establecer la normalidad de la estructura del cromosoma 15. También, pruebas con ultrasonidos en el feto no ofrecen ayuda para detectar anormalidades físicas relacionadas con AS dado que se espera que el feto afectado este bien formado. El volumen de líquido amniótico y los niveles de la alfa-feto-proteína también parecen normales.
Debido a las complejidades de evaluar riesgo de recurrencia, se aconseja la realización de un consejo genético hecho por un experto familiarizado con AS.
Reconocimientos:
Este documento ha sido desarrollado por la Angelman Syndrome Foundation con la ayuda de la
Raymond C. Philips Unit, Division of Genetics, Department of Pediatrics, University
of Florida.
- Angelman, H. "Puppet" children: A report on three cases. Dev Med Child Neurol. l965;7:681-688.
- Bower BD, Jeavons PM. The "happy puppet" syndrome. Arch Dis Child. 1967;42:298-302.
- Berg, JM and Pakula, Z. Angelman's ("happy puppet") syndrome. Am J Dis Child. 1972; 123:72-74.
- Berggreen, S. "Happy puppet" syndrome. Ugeskr Laeger. 1972; 134:1174.
- Kibel MA, Burness FR, The "happy puppet" syndrome. Centr Afr J Med. 1973; 19:91-93.
- Mayo 0, Nelson MM, Townsend, HRA. Three more "happy puppets". Dev Med Child Neural. 1973;1S:63-74.
- Moore JR, Jeavons PM. The "happy puppet" syndrome: Two new cases and a review of five previous cases. Neuropaediatrie. 1973;4: 172-179.
- Elian M. Fourteen happy puppets. Clin Pediatr. 1975; 14:902-908.
- Pashayan H, Singer W, Dove C, Eisenberg E, Seto B. The Angelman syndrome in two brothers. Am J Med Genet. 1982;13:295-298.
- Williams CA, Frias IL. The Angelman ("happy puppet") syndrome. Am J Med Genet. 1982; 11:543-460.
- Clayton-Smith J and Pembrey ME. Angelman syndrome. J Med Genet. l992;29(6):412-415.
- Saitoh, S., Harada, N., Jinno, Y., et al. Molecular and clinical study of 61 Angelman syndrome patients. Am J Med Genet. 1994;52: 158-163.
- Zori RT, Hendrickson J, Woolven S, Whidden EM, Gray B, Williams CA. Angelman syndrome: clinical profile. J Child Neuro. 1992;7(3):279-280.
- Chan CTJ, Clayton-Smith J, Cheng XJ, et al. Molecular mechanisms in Angelman syndrome: a survey of 93 patients. J Med Genet. 1993;30:895-902.
- Buntinx IM, Ilunnekam RCM, Brouwer OF, Stroink H, Beuten J, Mangelsehots K, Fryns JP. Clinical profile of Angelman syndrome at different ages. Am J Med Genet. 1995;56: 176-183.
- Angelman H (1991): personal correspondence
- Williams CA, Angelman H, Clayton-Smith J, Driscoll DJ, Hendrickson JE, Knoll JHM, Magenis RE, Schinzel A, Wagstaff J, Whidden EM, Zori RT. Angelman syndrome: Consensus for diagnostic criteria. Am J Med Genet. 1995;56:237-238.
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genet. 1997;15:70-73
- Matsuura T, Sutcliffe JS, Fang P. et al. De novo truncations mutations in E6-AP ubiquitin-protein ligase gen (UBE3A) in Angelman syndrome. Nature Genet. 1997;15:74-77.
- Mitch WE, Goldberg AL. Mechanisms of Muscle Wasting The Role of the Ubiquitin-Proteasome Pathway. NEJM. 1996;335:58-64
- Saitoh S, Buiting K, Cassidy S, et al. Clinical Spectrum and Molecular Diagnosis of Angelman and prader-Willi Syndrome Patients With an Imprinting Mutation. Am J Med Genet. 1997;68:195-206
- Fryburg JS, Breg WR, Lindgren V. Diagnosis of Angelman Syndrome in infants. Am J Med Genet. 1991;38:58-64.
- Clayton-Smith, J. Clinical research on Angelman syndrome in the United Kingdom: observations on 82 affected individuals. Am J Med Genet. l993;46(1): 12-15.
- Bottani A, Robinson 'VP, DeLozier-Blanchet CD, et al. Angelman syndrome due to paternal uniparental disomy of chromosome 15: A milder phenotype? Am J Med Genet. 1994;51:35-40.
- Wagstaff J. Genetic and Clinical Studies of Angelman Syndrome. Angelman Syndrome Foundation Medial and Scientific Symposium. July 3, 1997, Seattle, Washington.
![]()
![]()