Estudio sobre las Capacidades de Comunicación Expresivas
en Nińos con Síndrome de Angelman
Robin L. Alvares, Ph. D.
Sharon F. Downing, M.S.
Northern Illinois University, DeKalb
From the American Journal of Speech-Language Pathology, May,
1998, Vol. 7, No. 2, pp14-24.
Received May 29, 1997
Accepted January 21, 1998
Contactar con el autor: Robin L. Alvares, PhD, Department of Communicative Disordres, Northern Illinois University, DeKalb, IL 60115
Palabras clave: Síndrome de Angelman, retraso, intervención preverbal.
RESUMEN
El Síndrome de Angelman (SA) se produce como consecuencia de la deleción parcial del cromosoma 15 (Knoll y colaboradores, 1989) y ocurre en, aproximadamente, 1 por cada 10.000 nacimientos vivos (Peterson, Brondum-Nielsen, Hansen, & Wulff, 1995). Los individuos con SA muestran un modelo de retraso de desarrollo que incluye, problemas con la alimentación durante la infancia, retraso en el desarrollo motor, movimientos atáxicos, convulsiones, de severo a profundo retraso mental, y una falta de expresión hablada (Wills, Zori y colaboradores, 1995). Los objetivos de este artículo son resumir investigación clínica sobre SA, proporcionar una descripción de las habilidades de comunicación de individuos con SA, y, por último, identificar estrategias y recursos para la intervención en el área de comunicación. Empieza repasando la literatura existente sobre las características clínicas de los individuos con SA, haciendo énfasis en las habilidades de comunicación. La segunda parte del artículo presenta los resultados obtenidos en un estudio de 20 familias de nińos con SA en las habilidades de comunicación expresivas. Se discuten las implicaciones de los resultados del estudio con relación a la literatura existente.
INTRODUCCION
El Síndrome de Angelman SA se produce como resultado de la deleción parcial o trastorno del cromosoma 15 (Knoll, Nicholls, Magenis, Graham, Lalande, & Latt, 1989). El Síndrome de Angelman SA se ha identificado, en USA, en más de 600 individuos (Fundación del Síndrome de Angelman, 1993). Este número puede parecer bajo, aunque recientes estimaciones hacen pensar en una proporción de incidencia de 1/10.000 nacimientos vivos, y muchos casos de SA pueden estar sin diagnosticar (Peterson, Brondum-Nielsen, Hansen, & Wulff, 1995).
Los individuos con SA exhiben un modelo de retraso de desarrollo característico. Algunos de los rasgos clínicos asociados con el SA son: problemas con alimentación durante la infancia, retrasos en el desarrollo motor, movimientos espásticos (atáxicos), convulsiones, hiperactividad, retraso mental severo o profundo, y falta de expresión verbal. Los rasgos craneo-faciales pueden incluir un aplanamiento del cráneo en la región o ccipital, boca ancha, lengua y mandíbula protuberantes, y dientes espaciados o irregulares. El noventa por ciento de nińos con SA tienen convulsiones antes de la edad de 4 ańos (Angelman, 1965; Clayton-Smith, 1992, 1993; Williams, Hendrickson, Whidden, & Bueher, 1993). Debido a los arranques de risa frecuentes, expresión facial sonriente y una forma de andar torpe con flexión de los codos, SA fue inicialmente llamado el síndrome de la muńeca feliz, por el Dr. Harry Angelman por quien el síndrome se llama (Angelman, 1965).
Aunque el síndrome se describió por primera vez en 1965 (Angelman, 1965), solo recientemente han sido estudiadas las implicaciones sobre el desarrollo en el SA. El principal interés de los logopedas, educadores y familias se centra en identificar por qué los individuos con SA desarrollan muy poca o ninguna comunicación verbal o vocalizaciones, y el uso de la comunicación alternativa (gestos, seńales manuales y fotografías) es menos sofisticado de lo que sería predecible por sus habilidades receptivas y cognoscitivas (Clayton-Smith, 1993; Jolleff & Ryan, 1993).
Este artículo resume la investigación en la comunicación en nińos con S.A., describe las habilidades de comunicación de nińos con S.A., proporciona sugerencias para la intervención en la comunicación. Empieza revisando la literatura existente en las características clínicas de individuos con SA poniendo especial énfasis en las habilidades de comunicación. Se discuten las implicaciones de los hallazgos del estudio relativas a la literatura existente. Específicamente, los nińos con SA que sirvieron como sujetos en el presente estudio mostraron mayores diferencias en la comunicación expresiva que las descritas en estudios anteriores. El artículo concluye con estrategias para la intervención en la comunicación por personas con SA.
REVISION DE LA LITERATURA
Diagnóstico
En la mayoría de los casos, los individuos heredan 23 cromosomas de cada padre resultando 23 pares de cromosomas numerados del 1 al 23. cada cromosoma contiene dos brazos largos (el q brazo) y dos brazos cortos (el p brazo). Algunos síndromes, como el síndrome de Down (Trisomía 21), es el resultado de un cromosoma extra dado que el par 21 tiene tres cromosomas en lugar de el par usual. En el caso del síndromes como 'X-Frágil', o 'síndrome-4q', parte del brazo de un solo cromosoma puede estar borrado. Los síndromes también pueden ser el resultado de un defecto de "marcado" en un solo gen como ocurre en 'sickle cell anemia'. Un desorden en la información de un solo gen puede producir malfomaciones de desarrollo que son transmitidas cuando el gen se reproduce.
El Síndrome de Angelman es el resultado de procesos genéticos complejos que solo ahora están empezando a ser entendidos. Según las recientes investigaciones, hay cuatro maneras en las que el Síndrome de Angelman puede heredarse. Según Williams, Zori y colaboradores (1995) la causa más común del SA es una deleción grande en el brazo largo del cromosoma 15 en la región q-13 del alelo (parte del par cromosomico) heredado de la madre. La figura 1 es una ilustración de la región 15 del cromosoma donde la deleción ocurre. Si la deleción ocurre en el cromosoma heredado del padre, el nińo tendrá Síndrome de Prader-Willi, el cual tiene una secuencia diferente al Síndrome de Angelman (Knoll y colaboradores 1989). En aproximadamente el 2% de los casos hay un trastorno en el 'imprinting control center' del gen, el cual causa disfunción en la región del 15q11-13 (ver figura 1). El cinco por ciento de nińos con SA ha heredado ambos cromosomas 15 de su padre (Uniparental Disomía Paterna). Los nińos con uniparental disomía parecen tener manifestaciones menos severas del síndrome que los nińos con deleciones más grandes (Williams, Zori y colaboradores 1995). En el restante 20%, no hay ninguna evidencia visible de una deleción del cromosoma. En el momento de escribir este estudio, se han publicado estudios proponiendo que el gen responsable del SA, UBE3A, ha sido aislado (Kishino, Lalande, & Wagstaff, 1997 y Matsuura y colaboradores 1997); La comprobación genética rutinaria no puede descubrir el Síndrome de Angelman, y la prueba genética más exacta es el test de Fluorescencia in situ Hibridación (FISH).
Figura 1. Clases genéticas del Síndrome de Angelman.
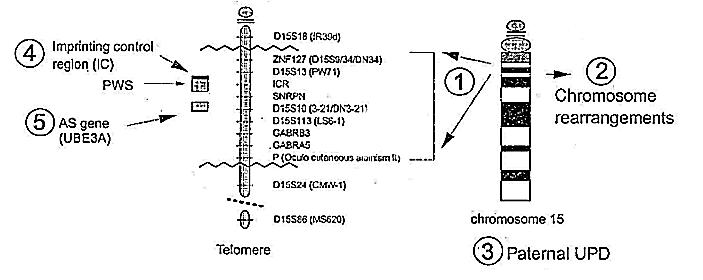
Ningún estudio epidemiológico a gran escala ha sido publicado. Se ha informado que la mayoría de los individuos identificados en América del Norte son blancos, sin embargo, casos de SA se han informado en grupos raciales y étnicos. No hay ningún dato acerca de la proporción entre varones y mujeres con SA. El riesgo de recurrencia es más alto en las familias en las que no se encuentra ninguna anormalidad en el cromosoma (Williams, Zori, y col. 1995).
El criterio de diagnóstico se ha establecido para confirmar el diagnóstico clínicamente en aproximadamente 80% de los casos (Williams, Angelman y col. 1995). mientras la comprobación genética es el medio más exacto de diagnosticar SA., aproximadamente el 20% de los casos se diagnostican solamente por las características clínicas del síndrome dado que no hay deleción visible . La Tabla I detalla las constantes características clínicas asociadas que se usan como criterio para el diagnóstico de SA. Electroencefalograma (EEG) es usado como apoyo del diagnóstico (Van Lierde, Atza, Giardino, y Viani, 1990; Williams, Angelman y col. 1995). Williams, Zori, y col. (1995) han propuesto que la hiperactividad puede ser un rasgo del síndrome más constante que la risa inadecuada y que la hiperactividad tiene un impacto negativo en el aprendizaje y en la comunicación social. Las características asociadas de particular importancia para logopedas son las características craniofaciales (lengua saliente, mandíbula prominente y boca ancha con los dientes espaciados), estrabismo (Schneider & Maino, 1993) dificultades con la alimentación y deglución en la infancia, habla con voz hueca y babeo.
Tabala I. Criterios clínicos consensuados para el Síndrome de Angelman
En el 100% de los casos diagnosticados
- Retraso del desarrollo: funcionalmente severo
- Deterioro del lenguaje: uso mínimo o nulo de palabras; las habilidades de comunicación receptivas y no verbales mayores que las verbales.
- Movimiento o desorden del equilibrio: normalmente andares atáxicos y/o movimiento trémulo de miembros.
- Comportamiento singular: Cualquier combinación de risa/sonrisa frecuente; la aparente conducta feliz; personalidad fácilmente excitable, a menudo con aleteo de manos; conducta hipermotorica, mantenimiento de la atención por periodos cortos.
Ocurrente frecuentemente (más del 80% de casos diagnosticados)
- Retrasado, desproporcionado crecimiento del perímetro cranioencefálico.
- EEG anormal, modelo característico.
Asociado (20% - 80% de casos diagnosticados)
- Aplanamiento de nuca. Aplanamiento de la fontanela.
- Lengua saliente, mandíbula prominente, boca ancha.
- Frecuente babeo, excesivas conductas de mordisqueo.
- Hipopigmentación en piel, pelo y ojos (solo en casos de deleción)
- Hiperactividad de las extremedidades inferiores y profundos reflejos de tendones.
- Levantada y flexionada la posición de los brazos especialmente durante la marcha.
- Perturbaciones del sueńo, atracción hasta la fascinación por el agua.
De: De Williams, Angelman y col. (1995). Síndrome de Angelman: Acuerdo consensuado para el diagnóstico. American Journal of Medical Genetics, 56, p. 238. (reimpreso con permiso).
Gran parte del retraso del desarrollo observado en individuos con SA puede ser atribuido a anormalidades neurológicas; ha habido sólo unos casos, sin embargo, en que el cerebro de individuos con SA ha estado disponible para estudios "post-mortem". Algunas anormalidades neurológicas han sido informadas incluyendo hiperplasia cerebral (excesivo crecimiento del tejido) (Frias, King & Williams, 1982) e hipoplasia unilateral temporal (pérdida de tejido) (Van-Lierde y col. 1990). otras diferencias neurológicas observadas incluyen disminución de la mielinización y adelgazamiento del cuerpo calloso (Buntix, Hunnekam, Brower, Stroink, Beuten, Mangelschots, & Fryns, 1995; Zori, Hendrickson, Woolven, Whidden, Gray & Williams, 1992). La mayoría de los individuos con SA tienen convulsiones (aproximadamente el 90%), y las convulsiones generalmente ocurren antes de los 3 ańos. Las convulsiones son generalmente de tipo motórico y afectan a todo el cuerpo, sin embargo, los individuos con SA pueden experimentar otros tipos de convulsiones. Debido a las convulsiones, muchos nińos reciben medicación anticonvulsiva, y no está claro como las medicaciones pueden afectar al aprendizaje de los nińos con SA.
El Síndrome de Angelman es a menudo no reconocible en el nacimiento, y a menudo el diagnóstico ocurre entre los 3 y los 7 ańos de edad, cuando las características del comportamiento se vuelven más aparentes (Williams y col. 1995). Las más sobresalientes características son severo retraso del desarrollo, falta del lenguaje, risa frecuente, lengua saliente, rigidez, andares inestables, convulsiones, hiperactividad e hipopigmentación de la piel. El Síndrome de Angelman puede no ser reconocido hasta que el nińo es evaluado en la infancia. Es importante para el logopeda estar familiarizado con las características clínicas del Síndrome de Angelman porque ellos pueden estar involucrados en la identificación inicial del síndrome.
Estudios de Habilidades de Comunicación en Individuos con Síndrome de Angelman.
Existen estudios que informan que unos pocos individuos con AS desarrollan un lenguaje utilizable, y se han informado retrasos en la habilidad para usar otras formas de comunicación expresiva tales como signos, gestos, etc. (Clayton-Smith, 1993; Jolleff & Ryan, 1993; Penner, Johnston, Faircloth, Irish, & Williams, 1993) Factores que se han propuesto para explicar el retraso expresivo incluyen retrasos cognoscitivos y receptivos, dificultades orales motóricas, pobre habilidad para la comunicación social y diferencias entre los sujetos seleccionados y los procedimientos de la recogida de datos. La siguiente sección presenta la descripción de las habilidades de comunicación en individuos con SA y las hipótesis con respecto a la naturaleza subyacente de los déficits expresivos observados en SA.
Factores cognoscitivos y Receptivos.Mucho del retraso en la comunicación expresiva observada en individuos con SA pueden ser atribuidos al retraso mental (Williams, Zori y col. 1995). Sin embargo, algunos estudios sugieren que el retraso cognoscitivo no es suficiente para el retraso expresivo del lenguaje asociado con SA.
Un estudio de las habilidades de la comunicación de un grupo de nińos con SA en el Reino Unido fue dirigido por Jolleff y Ryan (1993). Ellos evaluaron a 11 nińos con SA entre las edades de 2 ańos y 5 meses y 15 ańos y 3 meses, incluyendo un grupo de dos y un grupo de 3 hermanos. Los investigadores usaron dos medidas de comunicación para comparar la expresión hablada y la comprensión y las habilidades no-habladas expresivas y receptivas. La Escala Receptivas-Expresivas Lenguaje Emergente "The Receptive-Expressive Emergent Language Scale (REEL) (Bzoch & League, 1971) fue usada para obtener la puntuación de edad-equivalente para la comprensión del lenguaje hablado y las vocalizaciones expresivas y lenguaje. Una modificación de la Escala Preverbal de Comunicación "Preverbal Communication Scale (PVCS) (Kiernan & Reid, 1987) fue usada para evaluar las habilidades no-verbales receptivas y expresivas. La PVCS evalúa los tipos de gestos usados y entendidos por los individuos preverbales.
Usando el REEL, la edad-equivalente más alta para el lenguaje receptivo fue de 22 meses (rango 9 - 22 meses) El rango de puntuación para la comunicación expresiva fue menor (6 - 14 meses) y el lenguaje expresivo más alto fue de 14 meses de edad. La puntuación en lenguaje expresivo más alta fue asociada con los sujetos más mayores quienes eran también miembros de grupos de hermanos. Edades equivalentes para comunicación expresiva fueron menores que la comunicación receptiva en todos pero en un sujeto, y el hueco entre expresivo y receptivo en edades-equivalentes fue de 0 a 11 meses.
En el PVCS, los sujetos demostraron la habilidad de entender gestos simples (mano abierta para pedir y parecían mirar donde alguien seńalaba) pero hubo dificultades de imitación o utilización de gestos expresivos. Los cinco nińos de los dos grupos de hermanos fueron capaces de imitar actos motóricos simples (aplaudir, seńalar con la mano) pero los otros nińos no imitaron. Los autores pensaban que esto limitaba la utilización en la infancia de lenguaje de signos para la comunicación expresiva. La mayoría de los nińos usaron gestos ayudados por el contacto físico con el referente o interactuante (empujando una cosa no deseada o cogiendo la mano del adulto para situar la cosa en un sitio deseado). Los gestos de contacto son mentalmente menos sofisticados que los gestos manuales (por ejemplo seńalar, decir que no con la cabeza), lo cual no requiere un contacto físico con el objeto o el interactuante (McClean, McClean, Brady, & Etter, 1991; Wernes & Kaplan, 1963).
Los autores concluyeron que los retrasos expresivos exhibidos por nińos con SA podían no ser considerados solo debido a un retraso en el desarrollo mental porque habilidades expresivas no se correspondían con las habilidades receptivas. Ellos sugirieron posibles desconocidos mecanismos para el hueco entre habilidades receptivas y lenguaje expresivo: (a) dificultades motóricas para el acto de hablar, (b) problemas fundamentales en el uso de las comunicación para propósitos sociales, or (c) habilidades en comunicación expresiva en SA mejorando como una función bastante más que como un resultado del desarrollo de las habilidades receptivas o cognoscitivas.
Penner y col. (1993) evaluaron la comunicación cognoscitiva y habilidades motoricas de 7 adultos con SA. Todos los sujetos estaban institucionalizados y habían sido diagnosticados como profundamente retardados. Los investigadores encontraron que todos los sujetos funcionaban dentro del periodo sensoricomotor cognoscitivo registrado entre las edades de 2 - 6. Como Jolleff y Ryan (1993), Penner y col. concluyeron que los sujetos tenían dificultad imitativa gestual, incluso cuando ellos hacen gestos del repertorio de los sujetos. Los sujetos tendían a depender más del contacto más que de los gestos seńalados. Ninguno de los sujetos usaban comunicación simbólica, tales como lenguaje de signos o lenguaje mental (speech though) muchos usaron gestos usados como dar, manipulación física o intercambio de miradas entre el destinatario y el referente. Penner y col. concluyeron que hay variación en las habilidades de comunicación entre individuos con SA, incluso entre individuos clasificados como profundamente retrasados. Like Jolleff y Ryan (1993), los autores concluyeron que la falta de desarrollo cognoscitivo no se puede contar solamente por la falta del desarrollo del lenguaje.
Más investigaciones son necesarias para entender claramente la relación existente entre habilidades cognoscitivas y comunicación en nińos con SA. Hasta el presente, ha habido pocas evaluaciones sistemáticas de los diferentes aspectos de cognición, tales como atención, memoria y conducta simbólica. Williams, Zori y col. (1995) han especulado que el déficit de atención asociado con el desorden, combinado con los déficits motóricos, hiperatividad y pobres habilidades expresivas, hacen difícil de conseguir un perfil exacto de la función del desarrollo mental. Sin embargo, ellos especulan que algunas de las variables vistas en el síndrome se relacionan con la hiperactividad y que los nińos con mejores habilidades de atención dan mejor puntuación en las pruebas del retraso moderado.
Factores Orales Motóricos.Varios autores han sugerido que el retraso expresivo visto en individuos con SA pueden ser atribuidos parcialmente a las diferencias estructurales y a la disfunción en el mecanismo oral (Jolleff & Ryan, 1993; Frias, King, & Williams, 1982; Penner y col. 1993; Williams, Zori y col. 1995). Estos autores han informado varios problemas motóricos orales asociados con SA. Nińos con SA tienen dificultades de coordinación para succionar/tragar y tienen la lengua fuera que hace el amamantamiento difícil. muchos nińos con SA son alimentados con biberón, pero algunos requieren procedimientos quirúrgicos tales como una 'gastrostomy'. El desarrollo vocal temprano está caracterizado por la disminución en el llanto, balbuceo y arrullo. Una sola palabra puede desarrollarse entre los 10 y 18 meses, pero no es usada significativamente. En la nińez temprana se desarrollan otras conductas orales tales como mordisquear (llevarse todo a la boca) y babear y estas conductas pueden mantenerse hasta la madurez.
En su estudio, Penner y col. (1993) completaron exámenes orales motóricos a sus 7 sujetos adultos. La evaluación de la estructura y función del mecanismo oral visto en todos los sujetos mostraban retracción medio-facial con relativo prognatismo y dientes espaciados. otras diferencias craniofaciales fueron obervadas, tales como 'macrostomia' (excesiva boca ancha), y todos los sujetos tenian una boca abierta en reposo. Problemas de alimentación y babeo fueron observados en 6 de los 7 sujetos, y tuvieron conductas orales no adaptadas como mordisquear (llevarse todo a la boca) de objetos y manos, rumiar y malacia.
Frias, King y Williams (1982) informaron que el prognatismo observado en nińos con SA pueden ser un efecto secundario del excesivo mordisqueo y llevarse todo a la boca. los nińos que han sido estudiados tuvieron una medida normal de mandíbulas, pero las mandíbulas tenían una orientación delantera y ascendente. La orientación de la mandíbula, combinada con la retracción medio-facial y una pequeńa base craneal contribuyen all prognatismo observado en SA.
Factores de Comunicación Social. Se ha encontrado en la mayoría de los estudios realizados de individuos con SA una dificultad en la utilización de cualquier forma de comunicación social. Sin embargo no está claro cuánto de este retraso fue el resultado de factores relacionados con la experiencia y cuánto de características propias del síndrome. En el estudio de Jolleff y Ryan, 5 de los nińos en grupos de hermanos fueron los únicos sujetos que usaron gestos para comunicarse sin embargo muchos de los nińos habían tenido entrenamiento en la utilización del lenguaje de signos. Ellos informaron que sus sujetos nunca imitaron durante los ensayos clínicos, y sus sujetos parecían usar gestos, sobre todo, para la regulación de conductas (pedir) aunque ellos no trataban directamente las funciones de comunicación. Penner y col. (1993) también encontraron que los intentos de comunicación usados por estos sujetos fueron exclusivamente para pedir objetos o acciones y en ninguno de los sujetos se observó comentarios o protestas; sin embargo algunos sujetos demostraron la capacidad de unirse al grupo, interactuar y rechazar. Solo un sujeto se comprometió en una actividad recíproca de toma y daca. Se supone que los sujetos que tienen pobres habilidades sociales tienen limitadas sus habilidades para aprender el lenguaje debido a que tienen disminuidas las oportunidades para las interacciones comunicativas significantes.
Selección de sujetos y Factores de Recogida de Datos.
Clayton-Smith (1993) describieron el desarrollo mental y las características clínicas de SA en 82 pacientes con edades comprendidas entre los 17 meses y los 26 ańos en el Reino Unido. El próposito del estudio fue identificar desde el punto de vista físico y de comportamiento las características asociadas con la deleción del cromosoma y no fueron específicamente diseńadas para dirigirse al área de comunicación. La información se obtuvo a través de entrevistas a los cuidadores, y no hubo una valoración formal o informal de comunicación. Sin embargo el estudio de Clayton-Smith es interesante porque los hallazgos sugieren que algunos individuos con SA tienen más sofisticadas capacidades expresivas que otros de los sujetos descritos por Jolleff y Ryan (1993) y Penner y col. (1993). Like Jolleff and Ryan, Clayton-Smith encontraron unos pocos individuos con SA que usaban lenguaje para comunicarse. Ningún sujeto habló más de 6 palabras reconocibles y el 30% no tenían lenguaje. Sin embargo, el 20% de los sujetos eran capaces de utilizar un lenguaje de signos limitado para comunicar, algunos usaban gestos incluido seńalar con el dedo, empujar o tirar, mientras otros usaban tableros de comunicación para comunicarse. Signos y tableros son formas más avanzadas y mentalmente más desarrolladas de comunicación, sugieren que algunos individuos con SA pueden tener una mayor habilidad para usar la comunicación simbólica que los informes anteriores han reportado.
Las diferencias en los hallazgos obtenidos por estudios experimentales de Jolleff y Ryan (1993) y Penner y col. (1993) y el estudio de Clayton-Smith (1993) pueden, en parte, ser explicados por las diferencias en los métodos de recogida de datos. Jojeff y Ryan obtuvieron las conductas gestuales en un emplazamiento clínico considerando que Clayton-Smith entrevistaron a las familias de la mayoría de los sujetos en sus propias casas. No está claro en el estudio de Jolleff y Ryan que los individuos a los que se les aplicó el PVCS y REEL conociesen a los sujetos. Si individuos con SA tienen dificultades en utilizar la comunicación para propósitos sociales, es probable que su actuación sea mejor en un ambiente familiar más que en un emplazamiento poco familiar y con sus familias en lugar de con interlocutores no familiares (conocidos).
También es posible que estos primeros estudios hayan descrito un subgrupo de individuos con SA. Por ejemplo, Penner y col. (1993) los sujetos eran adultos que habían sido diagnosticados como profunda y mentalmente retardados y estaban institucionalizados. Jolleff y Ryan proporcionaron solo información de la historia de los casos y no dieron detalles acerca del tipo de intervenciones que los sujetos habían recibido. En contraste, los nińos con SA con quienes los autores de este estudio han experimentado fueron predominantemente nińos que vivían en sus hogares y habían tenido programas de intervención tempranos.
Resumen.
Los informes de Jolleff y Ryan (1993), Penner y col. (1993) y Williams, Zori y col. !995) y Clayton-Smith (1993) son bastantes consistentes en sus descripciones de las capacidades de comunicación de individuos con SA. Parece haber consenso entre los investigadores acerca de un retraso expresivo que no es solamente atribuible a retrasos cognoscitivos, y puede, en parte, ser relacionado con trastornos de los mecanismos orales. Further, Penner y Col. (1993) y Williams, Zory y col. (1995) suponen que ese déficit puede ser el resultado las limitadas oportunidades de interacción social.
Un Estudio de las Habilidades de Comunicación expresivas en nińos con Síndrome de Angelman.
Los autores de las observaciones clínicas e informes de las familias de los nińos con SA han indicado que los casos informados en la literatura existente no reflejan el rango de habilidades exhibidas por nińos con SA. Esto es de particular preocupación porque las metas y pronósticos estadísticos basados en expectativas derivadas de la literatura actual pueden ser inapropiadas y pueden infravalorar el potencial para la comunicación desarrollado por algunos individuos con SA.
Métodos
En un esfuerzo inicial por confirmar los informes paternos y las observaciones de los autores, fue desarrollado un estudio informal de habilidades de comunicación de nińos con SA. Dado que SA es un desorden bastante raro, el estudio fue distribuido vía Internet para tratar de investigar individuos de una más amplia área geográfica El estudio se limitó a las familias de individuos con SA que tenían acceso a Internet, y no todos quienes recibieron el informe vía Internet quisieron participar. Debe resaltarse que, como otros estudios de comunicación de las habilidades de individuos con SA, los resultados son extremadamente limitados por la propia selección de la muestra. Hay 600 casos de SA en Estados Unidos y un número desconocido internacionalmente; en el mejor de los casos, el estudio representó aproximadamente 2-3% de los casos conocidos de SA en Estados Unidos. Los resultados de esta limitada muestra no deben ser considerados como representativos de los individuos con SA pero son expuestos ante todo para generar discusión clínica y animar a futuras investigaciones. Substancialmente grandes estudios de comunicación serían necesarios para determinar que factores puede relacionarse con la variabilidad en las habilidades de comunicación observadas en los sujetos del presente estudio.
Un cuestionario fue desarrollado con el cual se obtuvo información sobre la salud, desarrollo, comunicación y satisfacción en general con los profesionales (ver Apéndice A). Los investigadores subscritos a la lista de Angelman a través del correo electrónico . Se envió una carta introductoria para ver el posible interés en participar. El estudio se envió vía Internet. A los participantes se les dio la opción de enviar el cuestionario a través del correo electrónico o rellenarlo y devolverlo a través del correo regular.
Resultados
Información descriptiva. Un total de 20 familias de nińos con SA completaron el estudio. Los participantes incluyen individuos que viven en Finlandia (5%, n=1), Canadá (15%, n=3) y Estados Unidos (80%, n=16). Cinco participantes eran padres, 12 eran madres, uno de los cuestionarios fue completado por ambos padres, uno por la abuela, y uno por el logopeda con el permiso de la familia. Las edades comprendidas fueron desde los 17 meses hasta los 13 ańos y cuatro meses con una media de edad de 6 ańos y un mes (ver Tabla 2). Siete eran nińos, 12 eran nińas y no se obtuvo información sobre el sexo de uno de los sujetos. Nueve nińos tenían un hermano, cinco nińos tenían dos hermanos, un nińo tenía tres hermanos, un nińo tenía 5 hermanos, y cuatro nińos no tenían hermanos. ninguna de las familias participantes respondió que tuviese más de un nińo con Síndrome de Angelman pero un nińo tenía una hermana mayor con espina bífida. Todos los nińos vivían con sus padres biológicos en sus casas.
Tabla 2. Información de Desarrollo |
|||||||
Sujeto |
Edad |
Sexo |
Diaagnóstico |
Edad del Diagnóstico |
Visión |
Edad en la que se sentaron sin apoyo/meses |
Edad en la que empezaron a caminar/meses |
1 |
1,5 |
M |
D |
1,2 |
E |
9 |
NA |
2 |
2,3 |
M |
N |
1,6 |
G |
8 |
NA |
3 |
3,6 |
M |
D |
1,8 |
E |
12 |
NA |
4 |
3,6 |
M |
D |
3,0 |
E |
8 |
24 |
5 |
4,1 |
V |
D |
4,0 |
N |
8 |
48 |
6 |
5,2 |
M |
D |
2,1 |
N |
20 |
NA |
7 |
5,3 |
M |
N |
5,0 |
O |
9 |
23 |
8 |
5,11 |
M |
N |
5,6 |
N |
- |
21 |
9 |
6,0 |
M |
U |
4,0 |
N |
24 |
36 |
10 |
6,0 |
M |
N |
5,0 |
N |
18 |
48 |
11 |
6,2 |
M |
D |
4,0 |
N |
8 |
48 |
12 |
6,9 |
V |
N |
6,0 |
N |
8 |
17 |
13 |
6,11 |
M |
D |
1,4 |
G |
25 |
33 |
14 |
7,0 |
V |
D |
3,0 |
E |
13 |
NA |
15 |
7,0 |
V |
D |
1,3 |
E |
30 |
40 |
16 |
7,0 |
V |
N |
2,6 |
N |
10 |
14 |
17 |
7,3 |
V |
D |
4,0 |
E,G |
24 |
54 |
18 |
8,2 |
M |
N |
6,0 |
G |
11 |
NA |
19 |
9,0 |
V |
U |
6,0 |
N |
10 |
30 |
20 |
13,4 |
- |
D |
- |
G |
- |
24 |
Nota: V = Varón; M = Mujeres; - = No responde; D = Deleción; U 0 Uniparental disomía; N = no visible deleción; E = Estrabismo; G = Gafas; A = Astigmatismo; O = Otros; N = No problemas; NA = No camina independientemente. |
|||||||
Historia Médica: Se presenta información con respecto a las historias médicas y de desarrollo de los nińos en la Tabla 2. Las edades de los diagnósticos fueron desde los 14 meses a los 6 ańos de edad, con uno de los participantes que omitió esta información (X = 3 ańos y 7 meses). El 75% de los nińos mostraron diferentes tipos de convulsiones. La mayoría de los nińos que tenía convulsiones estaban tomando como medicación depakine. Los padres no informaron acerca de problemas de audición en sus nińos, pero varios informaron sobre problemas de visión, incluyendo 25% ( n= 5) con estrabismo, 20% (n = 4) llevaban gafas, uno (5%) llevaba gafas y tenía astigmatismo, otro nińos con otros problemas de visión y uno (5%) con estrabismo y gafas.
El 65% ( n = 13) de los nińos habían conseguido andar independientemente. La edad en que los nińos iniciaron la marcha fue desde los 14 meses hasta los 54 meses, con una media de edad de 32 meses. El 35% ( n = 7) de los individuos que no andaban independientemente en edades desde los 17 meses hasta los 8 ańos y dos meses. Todos estos individuos que no andaban independientemente estaban andando con alguna forma de asistencia, con la excepción de un nińo de 17 meses de edad que se arrastrándose sobre su tripa y era capaz de soportar su propio peso cuando se le sostenía desde arriba. El noventa por ciento de los nińos se sentaba sin apoyo según los informes.
Fueron comunes los problemas con la alimentación, el 75% ( n = 15) de los participantes informaron que los nińos con SA tuvieron problemas con la alimentación incluyendo dificultades para mamar cuando eran pequeńos, reflujo, y preferencias y aversiones con la alimentación. Un nińo tenía un tubo de alimentación directa al estómago en el momento de realizar el estudio y no era capaz de tolerar líquidos oralmente.
comunicación. Se resumen modalidades y habilidades socio-comunicativas en las Tablas 3, 4 y 5. El uso de signos manuales y AAC ( Aumentativa/Alternativa Comunicación) varió ampliamente (Tabla 3). El 50% ( n = 10) de la muestra usaban alguna forma de signos comunicativos. De estos quienes utilizaban signos, 35% ( n = 7) usaban signos espontáneamente para comunicación funcional. El número de signos informados usados por los nińos con SA varían desde 2 hasta por encima de 200. Solo dos individuos usaban más de 10 signos, y solo un individuo usaba más de 200 signos y otro que usaba aproximadamente 40 signos. En general, los participantes informaron que estos signos eran aproximaciones y eran difíciles de entender debido a las limitaciones motóricas de los nińos. El 20% ( n = 4) de los nińos usaron signos para ganar la atención del interlocutor previamente a la utilización del lenguaje de signos. De acuerdo con las respuestas, el 55% ( n = 11) mantenían el adecuado contacto visual durante la comunicación con el interactuante. Tres de los participantes comunicaron que sus nińos eran muy sociables y mantenían un gran contacto visual. Cuando se comunicaban con sus nińos, el 60% ( n= 12) de los participantes informaron que ellos solamente usaban el lenguaje oral y solo un 30% ( n= 6) usaban signos al mismo tiempo que el lenguaje oral para comunicarse. Ninguno respondió que usase solo el lenguaje de signos y solamente dos participantes no facilitaron esta información.
Tabla 3. Uso de la comunicación no oral simbólica |
|||||||
Sujeto |
Edad |
Lenguaje signos |
Utiliza signos espontáneamente |
Numero de signos |
Intentaba conseguir atención previa |
La familia hace los signos al nińo |
Comunicación |
1 |
1,5 |
N |
-- |
-- |
-- |
N |
N |
2 |
2,3 |
S |
S |
9 |
N |
S |
F,V |
3 |
3,6 |
N |
-- |
-- |
-- |
S |
N |
4 |
3,6 |
N |
-- |
-- |
-- |
N |
N |
5 |
4,1 |
N |
-- |
-- |
-- |
-- |
F |
6 |
5,2 |
N |
-- |
-- |
-- |
S |
F |
7 |
5,3 |
S |
S |
40 |
S |
S |
N |
8 |
5,11 |
S |
S |
200 + |
S |
S |
F |
9 |
6,0 |
S |
S |
7 |
S |
S |
F,V |
10 |
6,0 |
N |
-- |
-- |
-- |
N |
F,V |
11 |
6,2 |
S |
S |
5 |
N |
N |
F |
12 |
6,9 |
S |
S |
8 |
N |
S |
V |
13 |
6,11 |
S |
S |
4 |
S |
S |
F,V |
14 |
7,0 |
N |
-- |
-- |
-- |
S |
V |
15 |
7,0 |
N |
-- |
-- |
-- |
N |
N |
16 |
7,0 |
N |
-- |
-- |
-- |
N |
N |
17 |
7,3 |
S |
N |
3 |
N |
S |
F |
18 |
8,2 |
N |
-- |
-- |
-- |
-- |
N |
19 |
9,0 |
S |
N |
7 |
N |
S |
V |
20 |
13,4 |
S |
N |
2 |
N |
S |
F,V |
Nota: S = Si; N = No; -- = No responde; F = Fotografías; V = Sistema de producción de voz |
|||||||
Sistemas de Comunicación Aumentativa/Alternativa (AAC) fueron usados por la mayoría de los individuos con SA. Fotografías fueron usadas solamente por el 25% ( n= 5) individuos. Aparatos de comunicación con voz fueron usados solamente por el 15% ( n = 3) de los nińos y junto con fotografías por el 30% ( n = 6 ) de los nińos. Treinta % ( n = 6 ) no usaron ninguna forma de comunicación aumentativa.
Los gestos eran comunmente usados por los nińos (Tabla 4). Cincuenta y cinco por ciento ( n = 11 ) seńalaban con el dedo para indicar necesidades o intereses y el 75% ( n = 15 ) para alcanzar los objetos deseados y el 75% ( n = 15 ) eran capaces de indicar si y no moviendo la cabeza. Se preguntó a los participantes si los nińos juntaban sus labios para soplar para determinar si los nińos estaban usando el gesto del soplado para representar la acción de soplar. Sin embargo, muchos participantes interpretaron esto como una pregunta sobre habilidades motoricas orales. Solo el 15% ( n = 3 ) de los participantes contestó que el nińo podía redondear los labios para soplar, la mayoría informó que problemas motóricos hacían esta actividad difícil para los nińos.
Tabla 4. Uso de Comunicación Gestual |
||||||||
Sujeto |
Edad |
Utilizan |
Seńala con el dedo |
Alcanzan |
Contacto físico |
Mueven cabeza para si/no |
Redondea los labios |
Mantiene |
1 |
1,5 |
S |
N |
S |
N |
N |
-- |
N |
2 |
2,3 |
S |
S |
N |
N |
S |
N |
S |
3 |
3,6 |
S |
S |
S |
N |
S |
S |
S |
4 |
3,6 |
N |
N |
N |
N |
S |
N |
S |
5 |
4,1 |
S |
S |
S |
N |
N |
S |
S |
6 |
5,2 |
N |
N |
A |
N |
S |
N |
N |
7 |
5,3 |
S |
S |
S |
S |
S |
N |
S |
8 |
5,11 |
S |
S |
S |
N |
S |
S |
S |
9 |
6,0 |
S |
S |
S |
S |
S |
N |
S |
10 |
6,0 |
S |
N |
S |
N |
N |
N |
N |
11 |
6,2 |
S |
S |
S |
N |
N |
S |
S |
12 |
6,9 |
S |
N |
A |
S |
S |
N |
S |
13 |
6,11 |
S |
S |
A |
N |
S |
N |
S |
14 |
7,0 |
S |
S |
A |
S |
S |
N |
S |
15 |
7,0 |
N |
M |
A |
S |
S |
N |
-- |
16 |
7,0 |
N |
N |
N |
S |
S |
N |
N |
17 |
7,3 |
S |
N |
S |
S |
S |
N |
N |
18 |
8,2 |
S |
N |
A |
S |
S |
N |
S |
19 |
9,0 |
N |
M |
A |
S |
S |
N |
-- |
20 |
13,4 |
S |
S |
N |
S |
S |
N |
S |
Nota: S = Si; N = No; -- = No responde; M = Toda la mano; A = Alcanza con la mano pero no abre/cierra. |
||||||||
Todos los nińos con SA vocalizaban y el 55% ( n = 11) de ellos usaban algo de lenguaje ( Tabla 5 ). La número de palabras fue desde una a 15, con una media de cinco palabras. Se informó que las palabras fueron generalmente aproximaciones y algunos nińos fueron no constantes en su producción. La mayoría de los participantes informaron atípica calidad de voz, y calidad de voz considerada como dura (10%, n = 2), gutural (35%, n = 7), dura y gutural (5%, n = 1), dura, nasal y gutural (10%, n = 2) o sonido normal (35%, n = 7). Un participante dijo "otros" describiendo la calidad de voz de su hija como "profunda". Todo los sujetos informaron risa, sin embargo, solo 11 sujetos (55%) informaron momentos de risa inapropiada.
Tabla 5. Habla y Vocalizaciones |
|||||
Sujeto |
Edad |
Vocalizaciones |
Nş palabras usadas |
Calidad de voz |
Risa Apropiada |
1 |
1,5 |
S |
0 |
S |
A |
2 |
2,3 |
S |
4 |
D |
A,I |
3 |
3,6 |
S |
0 |
S |
A,I |
4 |
3,6 |
S |
3 |
O |
A,I |
5 |
4,1 |
S |
15 |
D,N,G |
A |
6 |
5,2 |
S |
0 |
D,G |
A,I |
7 |
5,3 |
S |
4 |
S |
A,I |
8 |
5,11 |
S |
0 |
S |
A |
9 |
6,0 |
S |
5 |
S |
A |
10 |
6,0 |
S |
3 |
S |
A |
11 |
6,2 |
S |
-- |
G |
A,I |
12 |
6,9 |
S |
3 |
G |
A |
13 |
6,11 |
S |
6 |
D,N,G |
A |
14 |
7,0 |
S |
0 |
G |
A,I |
15 |
7,0 |
S |
-- |
S |
A,I |
16 |
7,0 |
S |
0 |
G |
A |
17 |
7,3 |
S |
2 |
G |
A,I |
18 |
8,2 |
S |
0 |
G |
A,I |
19 |
9,0 |
S |
6 |
G |
A |
20 |
13,4 |
S |
1 |
H |
A,I |
Nota: S = Si; N = No; --= No responde; D = Dura; N = Nasal; G = Gutural; S = Sonido normal; O = Otros; A = Apropiada risa; I = Inapropiada risa. |
|||||
Otro. Los participantes informaron unánimemente que a los profesionales les estaba faltando información sobre SA, aunque el 60% ( n = 12 ) indicaron que ellos estaban contentos con los servicios que ellos estaban recibiendo de los profesionales, logopedas. Algunos también expresaron descontento ya que el diagnóstico se retraso debido a la falta de conocimiento del médico que les trataba. Además, el 60% ( n = 12) de participantes informaron que ellos sentían que las habilidades de comunicación de su nińo estaban infravaloradas. Generalmente, la infravaloración estaba relacionada con la comunicación receptiva más que con la comunicación expresiva. El quince por ciento ( n = 3 ) de las habilidades de comunicación de los nińos estaba según informes recibidos de acuerdo con el ambiente, aunque muchas familias informaron que las habilidades de comunicación de sus nińos eran más pobres en escenarios poco familiares o con personas poco familiares. La mayoría de los nińos (65%) tenía algún tipo de comunicación simbólica ( n = 13) aunque se recibió poca información específica sobre la comunicación simbólica. El 95% ( n = 19 ) de los nińos había recibido intervención temprana ( servicios de desarrollo mental previos a la edad de 3 ańos).
DISCUSION E IMPLICACIONES CLINICAS
Los resultados de esta limitada muestra de nińos con SA sugieren que la mayoría de los nińos de la muestra encajan con los perfiles descritos en previos estudios con respecto a la historia de convulsiones, alcanzando hitos en su desarrollo, edad de identificación, dificultades orales motoricas y habilidades expresivas de comunicación. Sin embargo, había un rango de habilidad en capacidades de comunicación expresiva no informadas previamente en estudios de individuos con SA. Específicamente, parece haber un gran rango en el uso de formas simbólicas de comunicación y capacidades de comunicación 'dyadic' en algunos nińos con SA que previamente se habían reportado en la literatura. Estos hallazgos serán discutidos con respecto a estudios previos de individuos con SA y las implicaciones clínicas.
Formas de Comunicación. Los hallazgos del presente estudio son consistentes con estudios anteriores con respeto al uso limitado de la lengua hablada por la mayoría de los individuos con SA. Para los individuos que pueden hablar, el lenguaje es a menudo descrito como "impreciso" y "difícil de entender". Las descripciones anteriores de habilidades motóricas orales y hallazgos obtenidos por el presente estudio sugieren que las dificultades orales motóricas juegan un papel en el retraso del lenguaje observado en individuos con SA. Los participantes en el presente estudio informaron problemas con la alimentación y el tragar de sus nińos con SA, así como las diferencias de calidad de la voz. Estas características, junto con los problemas motóricos generales asociados con el síndrome, apuntan a dificultades motóricas orales como factores colaboradores en el retraso del lenguaje. Es interesante notar que los participantes en el presente estudio informaron que algunos de sus nińos usaban vocalizaciones con sentido. Usaron vocalizaciones solo, por ejemplo, para conseguir la atención del oyente (interactuante) y haciendo signos o acompańado por gestos como la repetición al mismo tiempo de la sílaba /A/ para conseguir un objeto deseado. Es interesante hacer notar que solo aproximadamente la mitad de los participantes informaron risa inapropiada que inicialmente era una de las características del "Síndrome de la muńeca feliz' por Angelman en 1965. Esto está de acuerdo con las descripción de Williams, Zori, y col. (1995).
Estudios previos de individuos con SA han informado el uso de gestos y muy limitado uso de signos manuales. La mayoría de los nińos en este estudio utilizaban gestos y signos limitados de comunicación; sin embargo dos sujetos tenían adquiridos más de 40 signos. Es interesante notar que aproximadamente la mitad de los nińos con SA estaban aprendiendo juego simbólico. Algunos detalles estuvieron disponibles sobre el juego simbólico mostrado por nińos con SA. Se informó que una nińa pretendía servir el café a su familia y pretendía ser un gato. Es interesante también, que este sujeto era quien usaba un gran número de signos manuales. Otro nińo pretendió dar un chupete a su muńeca. Considerando que aparece un rango considerable en la habilidad de usar comunicación manual entre los individuos con SA, gestos o signos parecen ser la modalidad expresiva preferida por la mayoría de los individuos con SA.
El potencial de uso de sistemas de comunicación manual depende de un número de factores, incluyendo la imitación, la habilidad cognitiva requerida en el uso de sistema de comunicación simbólica, adecuadas habilidades motóricas finas y gruesas, y oportunidades del uso de los signos en intercambios comunicativos. Parece que todos estos factores pueden contribuir en las dificultades en el uso del lenguaje de signos en los nińos con SA. Los signos de individuos con SA son, a menudo, descritos como imprecisos, posiblemente debido a las limitaciones motóricas finas y gruesas asociadas con el síndrome. Además, Penner y col. (1993) y Jolleff y Ryan (1993) encontraron que incluso con las habilidades sensomotóricas requeridas, algunos individuos con SA tenían dificultades para imitar gestos sugiriendo que algunos individuos con SA pueden tener dificultades en la utilización del lenguaje de signos debido a pobres habilidades imitativas motóricas. No puede darse como regla que la falta de exposición a los signos manuales dé como resultado un uso limitado de signos manuales por los individuos con SA. Solo aproximadamente la mitad de los participantes informaron un uso consistente del lenguaje de signos en comunicación con individuos con SA. La relativa contribución de cada una de esas limitaciones puede variar entre los individuos con SA; sin embargo, el hecho de que un pequeńo porcentaje de nińos diagnosticados con SA puedan adquirir un sustancial vocabulario de signos manuales indica que el uso de signos manuales para la comunicación puede ser una opción para muchos individuos con SA.
Algunos individuos con SA usaban un sistema de AAC (Alternativa/Aumentativa Comunicación) diferente del de lenguaje de signos. Hay poca información publicada sobre el uso de dispositivos para comunicación aumentativa de individuos con SA. Aunque el estudio de Clayton-Smith (1993) informó que algunos individuos con SA usaban comunicación aumentativa, ninguno de los sujetos en el estudio de Penner y col. (1993) ni en el estudio de Jolleff y Ryan (1993) se informó del uso de ninguna forma de dispositivo para el lenguaje aumentativo. Los autores del estudio encontraron que aproximadamente la mitad de los sujetos usaron algun tipo de AAC (Alternativa/Aumentativa Comunicación). El estudio de Amstrong (1992) presenta un caso ilustrativo de como un joven chico con SA fue entrenado para usar un dispositivo de voz (Wolf). Ella describe como la introducción temprana de AAC (Alternativa/Aumentativa Comunicación) fue un éxito con el cliente, N.M. se volvió más independiente con su juego y menos dependiente de su familia para interpretar sus esfuerzos comunicativos. Investigaciones más extensas son necesarias para determinar como individuos con SA pueden efectivamente usar AAC (Alternativa/aumentativa Comunicación). Sin embargo, si el problema primario de la falta de habla en el SA estuviese en problemas motóricos, entonces algunos nińos con SA podrían ser buenos candidatos para dispositivos de AAC (Alternativa/aumentativa Comunicación).
Comunicación Receptiva. Los estudios de comunicación en individuos con SA se han focalizado en habilidades de comunicación expresiva, y el presente estudio no se dirigió a habilidades receptivas. Es poco conocido el rango de habilidades receptivas en individuos con SA, y si individuos con SA muestran preferencias por ciertas modalidades receptivas (signos en oposición al habla).
Es interesante que ninguno de los padres informaron problemas en la audición con sus nińos. Uno de los autores se encontró con un número de familias de individuos con SA, y casi todas las familias de padres informaron que sus hijos tenían otitis media crónica.
Es necesario extensas investigaciones sobre la otitis media en SA así como el mecanismo fisiológico por el cual se llega a la otitis media crónica. La potencial contribución de la otitis media en las dificultades de comunicación en individuos con SA no debe ser pasada por alto.
Habilidades de Comunicación. Estudios previos de individuos con SA han informado pobre uso de las formas expresivas de comunicación en los intercambios de participación comunicativa. El presente estudio encontró que las habilidades de comunicación social en algunos nińos con SA son mejores que las informadas anteriormente. por ejemplo, algunos de los nińos conseguían previamente la atención del oyente para pedir o comentar. Sin embargo, la mayoría de los participantes informaron que las habilidades de sus nińos eran pobres con interlocutores no familiares y en ambientes no familiares. Esto puede explicar parcialmente el uso limitado de comunicación para propósitos sociales informado en estudios anteriores. La habilidad de participar en intercambios comunicativos varía entre los nińos con SA, y algunos nińos desarrollan la habilidad de usar formas comunicativas para expresar una variedad de intentos comunicativos y participar en interacciones sociales.
Implicaciones para la Intervención en Nińos con S.A.
Aunque los resultados del estudio describen como debe ser la interpretación conservadora, los resultados obtenidos a través de este estudio generalmente apoyan los estudios anteriores de Clayton-Smith (1993), Jolleff y Ryan (1993) y Penner y Col. (1993). Sin embargo, han surgido varias diferencias. El rango de habilidades encontrado en un número pequeńo de nińos con SA en el presente estudio excede de los informados en previos estudios. Estos resultados sugieren que un pequeńo grupo de nińos con SA pueden tener más potencial para la comunicación social simbólica que lo informado anteriormente.. Las diferencias pueden ser por varias razones incluyendo consideraciones de la muestra, consideraciones contextuales y experiencia en los servicios de intervención tempranos. Un estudio extenso y documentación del rango de habilidades exhibidas por individuos con SA facilitaron el diagnóstico, valoración e intervención.
La principal preocupación para muchas familiares, profesores y logopedas es la opción de la modalidad en comunicación. Hay muchos criterios a considerar, incluyendo el nivel individual del desarrollo simbólico, visión y habilidades motóricas, la disponibilidad de compańeros para la comunicación y preferencias familiares. Las familias y médicos tienen un número de opciones, y los médicos y las familias deben trabajar juntos para desarrollar sistemas de comunicación que sean al tiempo prácticos y útiles. En la actualidad, parece que la mayoría de los individuos con SA no desarrollarán lenguaje oral o serán incapaces de depender de la lengua como su primer modo comunicativo. El logopeda puede estar involucrado en las metas de desarrollo oral motórico para mejorar la alimentación y la forma de tragar y mejorar el babeo. Sin embargo, para un muy limitado numero de individuos con SA, la mejora de la lengua puede ser una meta apropiada. Para los individuos de nivel presimbólico, la intervención debe focalizarse en el uso de gestos y vocalizaciones teniendo como meta final el uso de signos manuales y/o un sistema de comunicación sinmbólico. Algunos médicos y familias han informado éxitos con sistemas gráficos como un sistema de intercambio de pictogramas, tanto como el "Alpha Talker". Equipos interdisciplinares quienes escogen el uso dispositivos de AAC (Aumentativa Alternativa Comunicación) pueden aprovecharse ellos mismos de los considerables datos clínicos y científicos disponibles.
Los individuos con SA exhiben algunas características únicas, tales como dificultades en el uso de habla expresiva, pero la intervención para los individuos con SA no es tan diferente de la intervención de individuos con otras discapacidades. La intervención en la comunicación debe servir para mejorar el desarrollo de comunicación verticalmente ayudando al individuo con SA para que alcance los más altos niveles de desarrollo en la comunicación a lo largo de la etapa pre-simbólica hasta la simbólica.
Además, los médicos deben asistir a eso que Halliday (1975) describió como los aspectos horizontales de la comunicación: el uso de las formas de comunicación a través del rango de las funciones comunicativas o intentos. Para aprender el lenguaje, los individuos deben participar en intercambios comunicativos con una variedad de interlocutores (compańeros comunicativos). Participacion en 'dyadic' intercambios requiere una variedad de habilidades incluyendo iniciación, respuesta, y la expresion de una variedad de intentos comunicativos.. Para individuos con poca o ninguna comunicación intencional, las metas de comunicación iniciales deben enfocarse a encontrar el modo comunicativo preferido del individuo y desarrollar programas con los que incrementan la comunicación intencional (Alvares & Sternberg, 1994). Mientras los estudios anteriores de la comunicación de individuos con SA sugieren muy limitadas habilidades sociocomunicativas, el presente estudio y observaciones clínicas han mostrado que algunos individuos con SA demuestran considerables competencias comunicativas en el uso de seńales, vocalizaciones, signos, sistemas de Aumentativa Alternativa Comunicación e incluso habla. Independientemente de la modalidad comunicativa, la intervención debe enfocarse en aumentar el número de compańeros comunicativos y debe reforzar la habilidad de participar en intercambios comunicativos de incrementar la duración, y desarrollar modalidades con las cuales expresar una variedad de intentos comunicativos.
Los profesionales frustran a las familias por la falta de conocimientos acerca del SA, y algunos profesionales pueden estar frustrados por la falta de información disponible del SA. Profesionales y familias deben fijar objetivos realistas para el desarrollo de la comunicación, pero las objetivos no deben estar equivocados en la meta a conseguir. No hay suficiente conocimiento sobre SA para hacer una larga escala de conclusiones sobre el potencial para el desarrollo de la comunicación en individuos con SA. Los profesionales deben ser sensibles a la frustración de las familias de individuos con SA proporcionando servicios de intervención de comunicación. Debido a que hay pocos conocimientos sobre el síndrome, las familias no saben que esperar y son desanimados por las expectativas limitadas que los profesionales tienen acerca de sus nińos. Las familias de nińos con SA no son poco realistas en sus expectativas sobre sus nińos, sin embargo, ellos son conscientes que muchos de sus "ángeles" son capaces de sobrepasar las expectativas de desarrollo actuales.
RECONOCIMIENTO
Los autores desean agradecer a las familias de los nińos con síndrome de Angelman que han participado en el estudio, las críticas por sus comentarios útiles y a los Drs. Charles Williams y Daniel Harvey por su ayuda en la interpretación de la información genética. Agradecemos también a Shraron y Rick Mason por su dedicación, ayuda y ánimo. Este artículo está dedicado a la memoria del Dr. Harry S. Angelman quien falleció en el otońo de 1996.
REFERENCIAS
Alvares, R., & Sternberg, L. (1994). Facilitating communication and language development. In L. Sternberg (De.) Individuals with Profound Handicaps: Instructional and Assistive Strategies, pp. 193-230. Austin, TX: Pro-Ed.
Comunicación facilitada y desarrollo del lenguage. En L. Sternberg (De.) Individuos con Deficiencias Profundas:
Angelman, H. (1965). "Puppet" children, a report on three cases. Developmental Medicine and Child Neurology, 7, 681-688.
Angelman Syndrome Foundation (1993). Angelman Syndrome (pamphlet).
Armstrong, B.L. (1992). Angelman’s syndrome: augmentative/alternative communication. Communication Outlook, 14 (2) - 14-21.
Buntinx, I.M., Hunnekam, R.C.M., Brouwer, O.F. Stroink, H., Beuten, J., Mangelschots, K, & Fryns, J.P. (1995). clinical profile of Angelman syndrome at different ages. American Journal of Medical Genetics, 56, 176-183.
Bzoch, K.R. & League, R. (1971). Rreceptive-Expresive Emergent Language Test. Austin, TX: Pro-Ed.
Clayton-Smith, J. (1992). Angelman’s syndrome. Archives of Disease in Childhood, 67, 889-91.
Clayton-Smith, J. (1993). Clinical research on Angelman Syndrome in the United Kingdom: Observbations on 82 affected individuals. American Journal of Medical Genetics, 46, 12-15.
Frias, J.L., King, G.J. & Williams, C.A. (1982). Cephalomeric assessment of selected malformation syndromes. Birth Defects, 18, 139-150.
Halliday, M.A.K. (1975). Learning How to Mean: Explorations in the Development of Language. New York: Academic Press.
Jolleff, N., & Ryan, M.M. (1993). Communication development in Angelman’s Syndrome. Archives of Disease in Childhood, 69, 148-150.
Kiernan, C., & Reid, B. (1987). Preverbal Communication Scale. Windsor, United Kingdom: NFER-Nelson.
Kishino, T., Lalande, M. & Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome (letter). Nature Genetics, 15, 70-73.
Knoll, J.H., Nicholls, R.D., Magenis, R.E., Graham, J.M. Jr., Lalande, & Latt, S.A. (1989). Angelman and Prader-Willi syndromes share a common chromosome deletion but differ in parental origin of the deletion. American Journal of Medical Genetics, 32 (2), 285-290.
Matsuura, T., Sutcliffe, J.S., Frang, P., aljaard, R. Jiang, Y., Benton, C.S., Rommens, J.M. & Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome (letter). Nature Genetics, 15, 74-77.
McClean, J.E., McClean, L.S., Brady, N.C. & Etter, R. (1991). Communication profiles of two types of gestures using nonverbal persons with severe to profound mental retardation. Journal of Speech and Hearing Research, 34, 294-308.
Mundy, P., & Seibert, J. & Hogan, A. (1984). Relationship between sensorimotor and early communication abilities in developmentally delayed children. Merrill-Palmer Quarterly, 30 (1), 33-48.
Nicolich, L.M. (1977). Beyond sensorimotor intelligence: Assessment of symbolic maturity through analysis of pretend play. Merrill-Palmes Quarterly, 23, 89-99.
Penner, K.A., Johnston, J., Faircloth, B.H., Irish, P. & Williams, C.A. (1993). Communication, cognition, and social interaction in the Angelman Syndrome. American Journal of Medical genetics, 46, 34-39.
Pterson, K.A., Brondum-Nielsen, K., Hansen, L.K. & Wulff, K. (1995). clinical, cytogenetic, and molecular diagnosis of Angelman Syndrome: Estimated prevalence rate in a Danish country (letter to the editor). American Journal of Medical Genetics, 60, 261-262.
Scheneider, B.B. & Maino, D.M. (1993). Angelman Syndrome. Journal of the American Optometric Association, 64, 502-506.
Sternberg, L. (1990). The Sonoma Research Project: Discussions and conclusions. In L. Sternberg (De.), Functional Communication: Analyzing the Nonlinguistc Skills of Individuals with Severe or Profound Handicaps. New York: Springer-Verlag.
Stillman, R., & Battle, C. (1984). Developing prelanguage communication in the severely handicapped: an interpretation of the van Dijk method. Seminars in Speech and Language, 5 (3), 159-169.
Van-Lierde, A., Atza, M.G., Giardino, D. & Viani, F. (1990). Angelman’s Syndrome in the first year of life. Developmental Medicine and Child Neurology, 32, 1011-1016.
Werner, H., & Kaplan, B. (1963). Symbol Formation. New York: John Wiley & Sons.
Wetherby, A., Yonclas, D., & Bryan, A. (1989). Communicative profiles of preschool children with handicaps: Implications for early identification. Journal of Speech and Hearing Disorders, 54, 148-158.
Williams, C.A., Angelman, H., Clayton-Smith, J. Driscoll, D.J., Hendrickson, J.E., Knoll, J.H.M., Magenis, R.E., Schnizel, A., Wagstaff, J. Whidden, E.M. & Zori, R.T. (1995). Angelman syndrome: Consensus for diagnostic criteria. American Journal of Medical Genetics, 56, 237-238.
Williams, C.A., Frias, J.L. (1982). The Angelman ("happy puppet") syndrome. American Journal of Medical genetics, 11, 453-460.
Williams, C., Hendrickson, J., Whidden, E., & Bueher, B. (1993). Facts about Angelman Syndrome. Unpublished manuscript. Gainesville, Florida: Department of Pediatrics, University of Florida.
Williams, C.A., Zori, R.T., Hendrickson, J., Stalker, H., Marum, T., Whidden, E., & Driscol, D.J. (1995). Angelman syndrome. Current Problems in Pediatrics, 25, 216-31.
Zori, R.T., Hendrickson, J., Woolven, S., Whidden, E.M. Gray, B. & Williams, C.A. (1992). Angelman syndrome: Clinical profile. Journal of Child Neurology, 7, 279-280.